IPOFISITI
Le ipofisitI linfocitarie (LYH) sono una malattia autoimmune della ghiandola pituitaria (ipofisi) (1) che può compromettere la secrezione ormonale ipofisaria.
Si distinguono in ipofisiti primarie ed ipofisiti secondarie (Tabella 1).
L’ ipofisite primaria rappresenta un’entità patologica confinata esclusivamente alla regione ipofisaria a differenza delle forme secondarie in cui il processo infiammatorio è innescato da un preciso agente eziologico infettivo o è espressione di malattie generalizzate. Da un punto di vista anatomo-patologico si distinguono 5 tipi di ipofisite primaria: linfocitaria, granulomatosa, xantomatosa, xanto-granulomatosa, necrotizzante (2). E ‘ancora discusso se queste forme siano entità realmente distinte oppure espressioni diverse della stessa patologia e, dal momento che condividono alcune caratteristiche, alcuni autori hanno ipotizzato una comune patogenesi autoimmune (3).
L’ ipofisite autoimmune, o ipofisite linfocitaria (LYH), è una forma primaria di ipofisite, in cui, come già accennato, l’infiammazione si limita alla sola ghiandola ipofisaria. La forma generalmente identificata come LYH è caratterizzata da un esteso infiltrato linfo-plasmacellulare a carico della ghiandola ipofisaria. Si definisce adenoipofisite linfocitica (LAH) un processo infiammatorio nel quale la disposizione dell’infiltrato coinvolge solo l’ipofisi anteriore, mentre nell’infundibulo-neuro ipofisite linfocitaria (LINH) tale processo interessa sia l’infundibulo che il lobo posteriore dell’ipofisi; infine nell’infundibulo-panipofisite linfocitaria (LIPH) è presente un coinvolgimento globale del lobo anteriore, di quello posteriore e dell’ infudibulum (4,5).
Un caso di panipopituitarismo con infiltrazione linfoplasmacellulare pituitaria è stato descritto da Rapp e Pashkis nel 1953 (6), ma non fu classificato come patologia autoimmune in quanto il concetto di autoimmunità endocrina è stato introdotto qualche anno più tardi per la tiroidite di Hashimoto (7). Una patogenesi autoimmune per la LYH è stata suggerita anni dopo per la prima volta da Gaudie e Pinkerton (8) che descrissero la comparsa di amenorrea post-partum e ipotiroidismo in una giovane donna in seguito deceduta per crisi surrenalica acuta da iposurrenalismo secondario in corso di appendicectomia. L’autopsia evidenziò una massiva infiltrazione linfo-plasmacellulare a carico dell’ipofisi e della tiroide nonchè atrofia dei surreni. Dopo questa prima descrizione alcuni case report sono stati segnalati in letteratura, ma studi originali sono stati molto rari e generalmente di piccole dimensioni.
Nel corso degli ultimi anni il numero dei casi segnalati è notevolmente aumentato probabilmente grazie al miglioramento della diagnostica di imaging. Tuttavia al momento la LYH è ancora poco considerata e la sua reale incidenza e prevalenza è sottostimata (9). Il motivo di misdiagnosi è la variabilità della storia naturale della malattia, caratterizzata dalle differenti espressioni cliniche e dalle numerose modificazioni temporali delle caratteristiche morfologiche, cliniche, funzionali ed immunologiche.
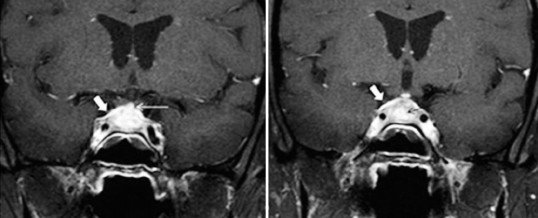
In alcune malattie endocrine la presenza di infiltrato linfocitario nelle ghiandole interessate può essere provata in modo relativamente facile a causa del semplice approccio alla ghiandola (10). Per quanto riguarda l’ ipofisite linfocitaria, i riscontri istopatologici, inizialmente su studi autoptici o bioptici post-chirurgici, sono stati ottenuti più recentemente mediante la biopsia pituitaria per via transfenoidale, ritenuta fino ad oggi il gold standard diagnostico per la LYH e le cui caratteristiche sono rappresentate da un processo infiammatorio con diffusa infiltrazione di linfociti, plasmacellule e macrofagi (9) (prenota valutazione neurochirurgica). L’ipotesi autoimmunitaria nella patogenesi dell’ipofisite linfocitaria oltre che dai dati istologici è sostenuta dal riscontro di caratteristiche alterazioni morfologiche di tale regione alla risonanza magnetica nucleare (RMN), nonché dall’associazione in più del 50% dei casi di ipofisite con altre malattie autoimmuni organo-specifiche e dal riscontro di anticorpi anti ipofisi (11).
Per quanto riguarda le caratteristiche morfologiche di imaging esse possono spesso essere suggestive di LYH; in particolare nei pazienti con sintomi o segni connessi all’allargamento dell’ipofisi, spesso la risonanza magnetica (RM) è in grado di differenziare l’ ipofisite linfocitaria dagli adenomi ipofisari, anche se talvolta il quadro tende a sovrapporsi. Nei pazienti con ipofisite linfocitaria, la RM ipofisaria di solito mostra un allargamento con estensione sovrasellare simmetrica della ghiandola che può spostare il chiasma ottico, mentre nei pazienti con adenoma ipofisario è presente un allargamento asimmetrico dell’ipofisi con deviazione del peduncolo (12).
La frequente associazione con altre malattie autoimmuni e la possibile presenza di altri autoanticorpi organo-specifici in pazienti con ipofisite linfocitaria è un ulteriore argomento a sostegno un coinvolgimento autoimmune in questa malattia (11). Questo è anche suffragato dalla buona risposta alla terapia immunosoppressiva e la comparsa di cicli di remissione e di recidiva frequentemente osservata nel corso della storia naturale della malattia, in analogia a quello descritto per altre malattie autoimmuni. In realtà, la ipofisite linfocitaria è frequentemente associata a malattie autoimmuni endocrine e non endocrine. L’associazione più comune è con la tiroidite di Hashimoto o morbo di Graves (13). Inoltre è stata descritta l’associazione con diabete insipido centrale, diabete mellito tipo 1, morbo di Addison, ipoparatiroidismo, gastrite cronica atrofica, anemia perniciosa; meno frequentemente la LYH può essere associata a lupus eritematoso sistemico, epatite autoimmune e cirrosi biliare primitiva. Si ricorda, infine, come alcune terapie oncologiche con immunomodulatori possano essere causa di ipofisite.
ANTICORPI ANTI IPOFISI
Anticorpi organo-specifici sono buoni indicatori di molte malattie endocrine autoimmuni. Gli anticorpi anti ipofisi (APA) non sono ancora considerati buoni marker di ipofisite linfocitaria a causa di varie difficoltà metodologiche, cliniche e interpretative. Infatti, l’impiego di metodi diversi nel rilevamento degli APA con conseguenti risultati contrastanti ha pregiudicato in passato la rilevanza clinica di questi anticorpi. Sulla base di queste differenze tra varie metodiche, quali l’immunoblotting e il i radioligand, di recente abbiamo effettuato una rivalutazione degli APA, mediante immunofluorescenza (Figura 1): la presenza degli APA è stata correlata a vari gradi di disfunzione ipofisaria; in particolare quando gli APA sono presenti a titolo elevato sembrano essere specifici marker di coinvolgimento autoimmune ipofisario in adulti e bambini con deficit di GH idiopatico e in adulti con ipogonadismo ipogonadotropo o deficit di ACTH idiopatici (14). La caratterizzazione successiva degli APA con tecnica di doppia immunofluorescenza ha permesso di dimostrare che le cellule somatotrope le gonadotrope e le cellule secernenti ACTH sono il bersaglio di questi anticorpi nei sieri di pazienti affetti dai sopradetti deficit. Allo scopo di migliorare ulteriormente il ruolo predittivo degli APA per l’insorgenza di un futuro ipopituitarismo, è stata effettuata una valutazione di questi anticorpi, non solo in base al titolo, ma anche al modello di immunostaining. Questa valutazione combinata di entrambi i parametri ha consentito di identificare pazienti a più alto rischio di disfunzione ipofisaria autoimmune, richiedendo pertanto una stretta sorveglianza ipofisaria allo scopo di rivelare una fase preclinica di ipopituitarismo ed eventualmente interrompere terapeuticamente la progressione della malattia clinicamente evidente (15).
STORIA NATURALE
In analogia ad altre malattie autoimmuni la storia naturale della LYH segue diverse fasi: l’infiammazione iniziale con allargamento della ghiandola corrisponde al periodo dei sintomi da effetto massa e, in particolare deficit ormonali subclinici che possono essere evidenziati da specifici test dinamici. La successiva distruzione dei tessuti e atrofia sono associati ad ipopituitarismo permanente (16). Tuttavia, in alcuni casi il decorso della malattia può essere piuttosto insidioso e sono stati riportati casi di recidiva/remissione di ipofisite linfocitaria (17). Infatti il processo infiammatorio può essere auto-limitante e il follow-up radiologico può mostrare una regressione nell’arco di circa 2 anni.
Può infine associarsi a diabete insipido centrale (CDI) completo o parziale.
DIAGNOSI
La LYH deve essere sospettata nei pazienti con iperprolattinemia, cefalea, alterazione del campo visivo, sintomi di ipopituitarismo soprattutto in donne in gravidanza e nel post-partum. Una diagnosi presunta di LYH può essere fatta oltre che con i dati clinici e di laboratorio anche mediante studi di imaging, ma la conferma richiede l’esame istopatologico, vale a dire la biopsia ipofisaria. Tuttavia, questa procedura è invasiva e non sempre fattibile. Inoltre, nonostante il recente sviluppo di tecniche sofisticate di imaging, la diagnosi di LYH alla risonanza magnetica resta problematica, poiché le conclusioni morfologiche di LYH su MRI possono sovrapporsi spesso con quelli di adenoma ipofisario (18).
Per questo motivo il rilevamento degli APA potrebbe essere utile per la diagnosi di LYH; in particolare la presenza degli APA a titoli elevati permette una diagnosi inequivocabile di LYH attiva, mentre il riscontro degli APA a bassi livelli potrebbe suggerire la diagnosi di possibile LYH di lunga durata. L’assenza degli APA non può escludere la possibilità di origine autoimmune della malattia, poiché gli autoanticorpi eventualmente presenti in passato, possono negativizzarsi nel tempo (prenota una visita endocrinologica).
TERAPIA
Prima di intraprendere qualsiasi terapia nella LYH, sono importanti due importanti considerazioni: in primo luogo, le diverse espressioni di questa malattia autoimmune richiedono differenti strategie terapeutiche; in secondo luogo, poiché si può osservare un’eventuale remissione spontanea durante la storia naturale della LYH, il miglioramento che si verifica dopo il trattamento chirurgico o medico può essere correlato a risoluzione spontanea piuttosto che al trattamento stesso. Per questo motivo un attento follow-up è consigliabile in pazienti senza sintomi di espansione extrasellare o di iposurrenalismo importante. Infatti, poiché la maggior parte dei decessi nei pazienti con LYH sono stati attribuiti a insufficienza surrenalica non trattata, la terapia sostitutiva con glucocorticoidi è essenziale nella fase acuta. La terapia comporta la sostituzione ormonale ipofisaria in caso di ipopituitarismo; poiché non è raro il riscontro di una risoluzione di alcuni o di tutti i deficit ipofisari, i pazienti che inizialmente sono stati sottoposti a trattamento sostitutivo devono essere ritestati successivamente per evitare terapie non più necessarie (17). Inoltre, in alcuni casi la terapia sostitutiva ormonale ipofisaria potrebbe agire come un “isohormonal terapy”, nel determinare il ripristino della funzione ipofisaria in LYH. In particolare, come dimostrato in altre malattie autoimmuni (19) anche nella LYH, quando la ghiandola pituitaria non è completamente e irreversibilmente distrutta, un feed-back di inibizione della funzione ipofisaria potrebbe diminuire l’esposizione di putativi autoantigeni ipofisari pituitari al ulteriori attacchi immuni. Glucocorticoidi o altri farmaci antinfiammatori ed immunosoppressori (metotrexate, ciclosporina A e azatioprina) sono stati indicati come trattamento medico, ma la loro efficacia a lungo termine deve essere ancora confermata (20,21). Il trattamento chirurgico per via transfenoidale, è utile per confermare la diagnosi, ma è anche molto efficace perché determina la decompressione della massa sellare (prenota una vista chirurgica) risolvendo prontamente la cefalea e i deficit visivi (prenota una vista oculistica) per cui è necessario nei pazienti con sintomi e/o segni di compressione grave (2). In alcuni casi la biopsia ipofisaria può essere sia diagnostica che terapeutica, perché dopo questa procedura un progressivo recupero della funzione pituitaria può essere osservato (11).
Prof.ssa Annamaria De Bellis
Bibliografia
1. Ezzat S and Josse. R Autoimmune Hypophysitis. In Volpè R (ed) Autoimmune Endocrinopathies. pp.337-348.Totowa New Jersey Humana Press Inc. 1999
2. Cheung CC, Ezzat S, Smyth HS, Asa SL. The spectrum and significance of primary hypophysitis. J Clin Endocrinol Metab 2001; 86: 1048-1053
3. Beressi N, Beressi JP, Cohen R, Modigliani E. Lymphocytic hypophysitis. A review of 145 cases. Ann Intern Med 1999; 150: 327-341
4. Vidal S, Rotondo F, Horvath E, Kovacs K, Scheithauer BW. Immunocytochemical localization of mast cells in lymphocytic hypophysitis. Am J Clin Pathol; 2002: 117 478-483
5. Tashiro T, Sano T, Xu B, Wakatsuki S, Kagawa N, Nishioka H, Yamada S, Kovacs K. Spectrum of different types of hypophysitis: a clinicopathologic study of hypophysitis in 31 cases. Endocr Pathol. 2002;13:183-195
6. Rapp JJ, Pashkis KE. Panhypopituitarism with idiopathic hypoparathyroidism. Ann Intern Med 1953; 39: 1103-1107
7. Roitt IM, Doniach D, Campbell PN, Hudson RV. Autoantibodies in Hashimoto’s disease. Lancet 1956; 2: 820-822
8. Goudie EB, Pinkerton PH. Anterior hypophysitis and Hashimoto’s disease in a young woman. J Pathol Bacteriol 1962; 83: 584-585
9. Bellastella A, Bizzarro A, Coronella C, Bellastella G, Sinisi AA, De Bellis A. Lymphocytic hypophysitis: a rare or underestimated disease? Eur J Endocrinol. 2003 149: 363-376
10. Witebsky E, Rose NR, Terplan K, Paine JR, Egan RW. Chronic thyroiditis and autoimmunization. JAMA 1957; 164: 1439-1447
11. De Bellis A, Bizzarro A, Bellastella A. Pituitary antibodies and lymphocytic hypophysitis. Best Pract Res Clin Endocrinol Metab. 2005;19:67-84
12. Levine SN, Benzel EC, Fowler MR, Shroyer JV 3rd, Mirfakhraee M. Lymphocytic adenohypophysitis: clinical, radiological, and magnetic resonance imaging characterization. Neurosurgery 1988; 22: 937-941
13. Barbaro D, Loni G. Lymphocytic hypophysitis and autoimmune thyroid disease. J Endocrinoll Invest 2000; 23: 339-340
14. De Bellis A, Pane E, Bellastella G, Sinisi AA, Colella C, Giordano R, Giavoli C, Lania A, Ambrosio MR, Di Somma C, Zatelli MC, Arvat E, Colao A, Bizzarro A, Bellastella A; Italian Autoimmune Hypophysitis Network Study. Detection of antipituitary and antihypothalamus antibodies to investigate the role of pituitary or hypothalamic autoimmunity in patients with selective idiopathic hypopituitarism. Clin Endocrinol 2011;75:361-6.
15. Bellastella G, Rotondi M, Pane E, Dello Iacovo A, Pirali B, Dalla Mora L, Falorni A, Sinisi AA, Bizzarro A, Colao A, Chiovato L, De Bellis A; Italian Autoimmune Hypophysitis Network Study. Predictive role of the immunostaining pattern of immunofluorescence and the titers of antipituitary antibodies at presentation for the occurrence of autoimmune hypopituitarism in patients with autoimmune polyendocrine syndromes over a five-year follow-up. J Clin Endocrinol Metab. 2010 95:3750-7
16. Goswami R, Kochupillai N, Crock PA, Jaleel A, Gupta N. Pituitary autoimmunity in patients with Sheehan’s syndrome. J Clin Endocrinol Metab. 2002; 87: 4137-4141
17. Matta MP, Kany M, Delisle MB, Lagarrigue J, Caron PH. A relapsing remitting lymphocytic hypophysitis. Pituitary 2002; 5: 37-44
18. Skandarajah A, Ng W, Gonzales M, Kaye A. Lymphocytic hypophysitis mimicking pituitary macroadenoma. J Clin Neurosci. 2002; 9: 586-589
19. De Bellis A, Colao A, Di Salle F, Muccitelli VI, Iorio S, Perrino S, Pivonello R, Coronella C, Bizzarro A, Lombardi G, Bellastella A. A longitudinal study of vasopressin cell antibodies, posterior pituitary function, and magnetic resonance imaging evaluations in subclinical autoimmune central diabetes insipidus. J Clin Endocrinol Metab 1999: 84: 3047-3051
20. Yamagami K, Yoshioka K, Sakai H, Fukumoto M, Yamakita T, Hosoi M et al. Treatment of lymphocytic hypophysitis by high-dose methylprednisolone pulse therapy. Internal Medicine 2003; 42: 168-173
21. Lecube A, Francisco G, Rodriguez D, Ortega A, Codina A, Hernandez C, Simo R. Lymphocytic hypophysitis successfully treated with azathioprine: first case report. J Neurol Neurosurg Psychiatry 2003; 74:1581-1583
APR
2012