NEOPLASIE ENDOCRINE MULTIPLE
Le Neoplasie Endocrine Multiple (MEN) si distinguono in MEN tipo 1 e MEN tipo 2, queste ultime a loro volta distinte in MEN 2A e MEN 2B.
Tutte le MEN condividono alcune caratteristiche.
In primo luogo, salvo alcune eccezioni, sono tutti tumori composti da cellule di origine neuroendocrine.
Altro aspetto comune è la caratteristica progressione istologica da iperplasia ad adenoma e, talora, a carcinoma.
Infine, tutte le MEN sono sindromi ereditarie con trasmissione autosomica dominante e ciò implica la necessità di una diagnosi precoce e dello studio dei familiari.
La MEN 1 (sindrome di Wermer) è caratterizzata da iperplasia delle paratiroidi, tumori del pancreas endocrino e tumori dell’ipofisi (3 P: parahtyroid, pancreas, pituitary).
La MEN 2 è detta anche sindrome di Sipple.
La MEN 2A è costituita da carcinoma midollare della tiroide, feocromocitoma e iperplasia/neoplasia delle paratiroidi. Una sua variante è il carcinoma midollare familiare (FMTC) della tiroide in cui l’unica manifestazione della sindrome è il tumore tiroideo.
La MEN 2B presenta carcinoma midollare della tiroide, feocromocitoma, neuromi multipli delle mucose, habitus marfanoide. (Prenota una visita endocrinologica).
EPIDEMIOLOGIA
La percentuale con cui si riscontrano tali neoplasie è variabile come si può osservare dalla seguente tabella:
MEN 1
Adenoma/iperplasia paratiroidi (>90%)
Tumori pancreas (80%)
Tumori ipofisi (25-50%)
Altri tumori (5-30%)
MEN 2A
Carcinoma midollare tiroide (90-100%)
Feocromocitoma (40-50%)
Adenoma/iperplasia paratiroidi (10-50%)
MEN 2B
Carcinoma midollare tiroide (90-100%)
Feocromocitoma (50-70%)
Neuromi mucosi (90%)
GENETICA
Il gene responsabile della MEN 1 è un oncosoppressore localizzato nel cromosoma 11 coinvolto nella crescita cellulare. La proteina prodotta da tale gene (gene MEN 1) è stata denominata menina. Affinché si manifesti la sindrome clinica è necessario che alla mutazione germinale si associ una mutazione somatica. Tali mutazioni del gene MEN 1 sono presenti in oltre il 90% delle famiglie affette. (Prenota un’analsi genetica per MEN).
Il gene responsabile della MEN 2, invece, è il protooncogenege RET localizzato nel cromosoma 10 che codifica per un recettore tirosin-chinasico. La mutazione di tale gene, a prescindere da dove si verifichi, causa uno stimolo continuo alla crescita cellulare e quindi lo sviluppo delle neoplasie tipiche dalla MEN 2. (Prenota una visita endocrinologica).
MEN 1
Il paziente che presenta una delle manifestazioni della MEN 1 deve esser indagato alla ricerca di eventuali altre lesioni e data l’ereditarietà devono essere indagati anche i familiari.
L’ereditarietà, infatti, ha un’alta penetranza ma una espressività variabile.
Pertanto l’anamnesi, la ricerca delle lesioni cutanee, la determinazione della calcemia, della glicemia, della prolattinemia, costituiscono una prima adeguata valutazione dei familiari di primo grado. La negatività dei risultati non esclude la possibilità futura della malattia, pertanto le indagini devono essere ripetute nel tempo.
Il paziente affetto da MEN può essere sottoposto ad analisi genetica del DNA estratto dai linfociti con ricerca di mutazioni del gene MEN 1. L’analisi andrà poi estesa ai familiari del paziente con lo scopo di arrivare ad una diagnosi precoce, prima che le manifestazioni cliniche diventino evidenti.
L’analisi genetica negativa di un componente di una famiglia nella quale sia presente una mutazione nota esclude che quel soggetto sia affetto con certezza pressoché assoluta.
Adenoma/iperplasia paratiroidi
L’iperparatiroidismo è la manifestazione clinica più frequente,causata dall’iperplasia di più paratiroidi.
A differenza dell’iperparatiroidismo dovuto ad adenoma vi è una mmagior sensibilità del tessuto iperplastico al calcio ed una minor percentuale di successo dopo l’intervento per la comparsa di recidive nel 50% dei casi.
I sintomi sono quelli dell’iperparatiroidismo e la terapia di scelta è quella chirurgica. Tuttavia le frequenti recidive possono richiedere plurimi interventi.
Tumori pancreatici
Rappresentano la seconda manifestazione clinica della MEN 1.
I tumori sono speso multicentrici e danno spesso metastasi. Fra questi ricordiamo il gastrinoma che causa la sindrome di Zollinger-Ellison e gli insulinomi. Più rari il glucanonoma, il VIP-oma ed il somatostatinoma.
Tumori ipofisari
I tumori ipofisari sono nella maggior parte dei casi prolattinomi (iperprolattinemia) e GH-omi (acromegalia). L’acromegalia nelle MEN 1 può esser causata, anche se raramente, da una secrezione ectopia di GHRH da parte di un tumore pancreatico. Più rari l’ACTH-oma (sindrome di Cushing).
Lesioni cutanee ed altri tumori
Nel 30% delle MEN 1 possono essere presenti lipomi sottocutanei, collagenomi, angiofibromi multipli ed altre neoplasie (adenomi del corticosurrene, lipomi, adenomi follicolari della tiroide etc.). (Prenota una visita endocrinologica).
MEN 2 A
Solitamente compare più precocemente il carcinoma midollare della tiroide, mentre l’interessamento delle altre ghiandole è più tardivo.
Pertanto la diagnosi è spesso basata sulla determinazione della calcitonina in condizioni basali o dopo stimolo (con pentagastrina o con il test al calcio) per determinare la presenza del carcinoma midollare della tiroide che si riscontra nel 100% dei casi. Questa valutazione deve riguardare sia il paziente che i suoi familiari, i quali vanno indagati sin dall’infanzia con lo scopo di porre una diagnosi precoce (la presenza di iperplasia delle cellule C va considerata una lesione precancerosa). Vanno dosate anche le catecolamine urinarie per la diagnosi di feocromocitoma. Anche la mutazione del gene RET va ricercata nel soggetto affetto e nei familiari. I familiari con assenza di mutazione RET non svilupperanno mai la malattia, in quelli con mutazione, invece, va effettuata una tiroidectomia preventiva prima che il carcinoma midollare risulti clinicamente evidente.
Carcinoma midollare tiroide
Il carcinoma midollare tiroide compare a partire da un quadro di iperplasia delle cellule C parafollicolari della tiroide.
In generale il carcinoma midollare tiroide si presenta con aggressività crescente dal carcinoma midollare familiare alla MEN 2A alla MEN 2B. In quest’ultima anche l’esordio è più precoce.
Feocromocitoma
Il feocromocitoma viene solitamente diagnosticato dopo il carcinoma midollare.
Il feocromocitoma origina a partire da un quadro di iperplasia sino alla comparsa di un feocromocitoma che può essere singolo o multifocale, monolaterale o più spesso bilaterale.
Una volta individuato il paziente deve essere sottoposto ad asportazione mediante surrenectomia monolaterale o bilaterale, in quanto alcuni studiosi sostengono che è molto probabile che si sviluppi un tumore della ghiandola controlaterale negli anni successivi.
Iperparatiroidismo
Si sviluppa nel 10-20 % dei casi in seguito ad un quadro di iperplasia più che a causa di un adenoma.
Lichen cutaneo amiloidosico
Si tratta di una lesione cutanea, pruriginosa, spesso iperpigmentata, localizzata in regione interscapolare.
Si riscontra in pochi pazienti ma è un segno precoce e patognomonico di MEN 2A. (Prenota una visita endocrinologica).
MEN 2 B
Carcinoma midollare tiroide
Il carcinoma midollare tiroide compare in tutti i pazienti ed ha un andamento altamente maligno.
Feocromocitoma
Il feocromocitoma in questo caso è quasi sempre bilaterale.
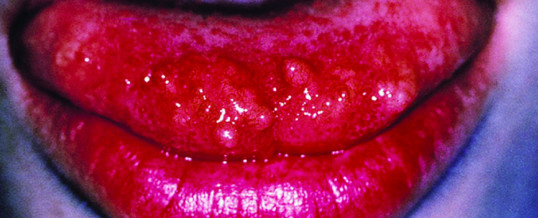
Neuromi mucosi multipli
Consistono in una crescita eccessiva di tessuto nervoso a livello delle mucose di lingua, labbra, cavo orale, congiuntive, cornea e tratto gastro-intestinale. In quest’ultimo caso si produce un quadro di megacolon che causa alterata motilità intestinale con diarrea alternata a stipsi.
I pazienti presentano un habitus marfanoide: i soggetti sono alti, magri, con petto scavato arti lunghi e sottili, ipotrofia muscolare. Sono assenti, tuttavia, le altre anomali tipiche della vera sindrome di Marfan.
Prenota una visita specialistica endocrinologica in merito a questo argomento.
Dott. Massimiliano Andrioli
Specialista in Endocrinologia e Malattie del Ricambio
Centro EndocrinologiaOggi, Roma
viale Somalia 33A, Roma
tel/fax 0686391386
cell 3337831426
Studio EndocrinologiaOggi, Lecce
via Ruffano 4, Casarano (Lecce)
tel/fax 0686391386
cell 3337831426
Bibliografia
Malattie del Sistema Endocrino e del metabolismo 2006, Faglia, Bek-Peccoz, McGraw-Hill.
SET
2011