TUMORI NEUROENDOCRINI
A metà degli anni 90 è stata proposta una revisione della vecchia e non chiara classificazione dei tumori neuroendocrini.
Nella nuova classificazione sono stati combinati gli aspetti patologici, di sede e funzionali secondo uno schema unificato applicabile a tutti i tumori endocrini GEP indipendentemente dalla sede e dal tipo di ormone prodotto.
Tale classificazione presenta indubbi vantaggi dal punto di vista classificativo ed ha implicazioni prognostiche e terapeutiche per cui essa è stata pressochè integralmente utilizzata dall’OMS nella recente riedizione della classificazione dei tumori endocrini (1).
Tale classificazione è basata sui seguenti criteri:
1. Nomenclatura
1 Limitazione (o abbandono secondo Cappella et al. 1995) del termine carcinoide. La definizione alternativa prescelta è quella di tumore o carcinoma endocrino, a seconda rispettivamente dell’assenza o della presenza dimostrata di malignità. In particolare la classificazione oggi utilizzata prevede le seguenti possibilità:
a. Tumori differenziati
2 Benigni
3 Indefiniti, benigni o potenzialmente maligni
b. Carcinomi ben differenziati (a basso grado di malignità)
c. Carcinomi scarsamente differenziati (ad alto grado di malignità).
4 Utilizzo combinato di dati anatomoclinici (sede, volume, documentata presenza di metastasi, presenza di angioinvasione) e funzionali (in particolare il tipo di secrezione ormonale e la presenza o meno di una sindrome clinica associata).
5 Suddivisione delle neoplasie per sede: stomaco, pancreas, duodeno, digiuno-ileo, appendice e colon-retto, oltre che polmone.
2. Caratteri istologici
3. Criteri prognostici
CLASSIFICAZIONE CLINICA
I tumori endocrini del tratto gastro-entero-pancreatico vengono clinicamente classificati in rapporto alla sede della neoplasia ed alla presenza o meno di sindrome associata. In particolare:
1. Tumori endocrini dello stomaco associati o meno a sindrome da carcinoide
2. Tumori endocrini del duodeno-digiuno prossimale associati o meno a sindrome
3. Tumori endocrini del digiuno distale-ileo-cieco associati o meno a sindrome da carcinoide
4. Tumori endocrini dell’appendice
5. Tumori endocrini del colon-retto
6. Tumori endocrini del pancreas
- INSULINOMA
- GASTRINOMA
- VIPOMA (SINDROME DI VERNER-MORRISON)
- GLUCAGONOMA
- CARCINOIDE
- NON ASSOCIATI A SINDROME
TUMORE ENDOCRINO DELLO STOMACO
Il tumore endocrino dello stomaco origina dalle cellule ECL presenti a livello della mucosa gastrica e costituisce circa il 6% dei tumori endocrini del tratto gastro-intestinale (2). Negli ultimi anni si è assistito ad un notevole aumento nella diagnosi di tali neoplasie. Tale incremento è verosimilmente da attribuire ad un maggiore utilizzo di procedure endoscopiche del tratto esofago-gastro-duodenale, ad un più frequente ricorso alla biopsia della mucosa gastrica e ad un maggiore ricorso, da parte dell’anatomo patologo, di specifiche tecniche di diagnosi immunoistochimica.
I tumori endocrini dello stomaco si suddividono in (3):
1 Tipo 1 o tumore associato a gastrite cronica atrofica (GCA)
2 Tipo 2 o tumore associato a Sindrome di Zollinger-Ellison in paziente con Neoplasia Endocrina Multipla di tipo 1 (ZES-MEN 1)
3 Tipo 3 o sporadico.
Tipo 1
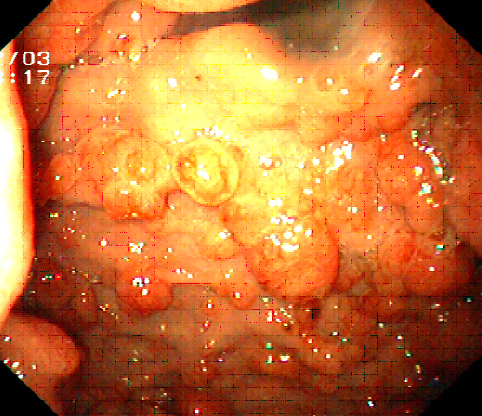
Il tumore endocrino dello stomaco associato a gastrite cronica atrofica (fig. 1) rappresenta la forma più frequente di neoplasia endocrina dello stomaco (prenota una visita gastroenterologica). Si tratta generalmente di lesioni multiple, di dimensioni inferiori a 1-2 cm, localizzate a livello della mucosa del corpo-fondo dello stomaco. Originano dalle cellule ECL dello stomaco secondo un progressivo passaggio partendo dalla semplice iperplasia, attraverso la displasia fino alla neoplasia vera e propria; presentano una crescita estremamente lenta e generalmente non determinano metastasi a distanza (4). Tale neoplasia risulta essere strettamente correlata alla gastrite cronica atrofica conseguete ad una patologia autoimmune (gastrite cronica atrofica di tipo A con positività degli anticorpi anti cellule parietali gastriche associata o meno ad anemia perniciosa) o sviluppatasi in seguito ad una infezione cronica da Helicobacter Pylori. Infatti l’atrofia gastrica è in grado di stimolare una iperincrezione di gastrina che, tramite il suo effetto trofico sulle cellule ECL dello stomaco, rappresenta lo stimolo primario allo sviluppo della iperplasia-displasia-neoplasia endocrina dello stomaco.
{kind=link}
Tipo 2
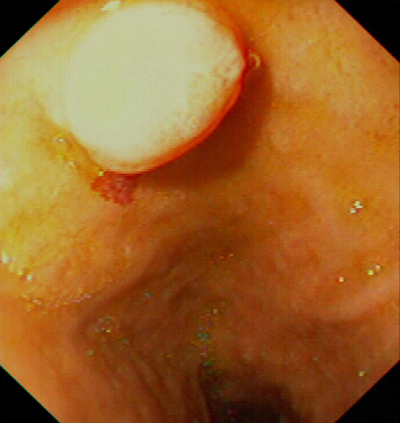
I tumori a cellule ECL di tipo 2 (fig. 2) sono rari, costituiscono appena il 6% di una larga serie recentemente studiata (4). Tali tumori generalmente insorgono in pazienti adulti senza prevalenza di sesso. Gli ECL-omi gastrici sono riportati nel 13-30% dei pazienti con ZES-MEN 1, nello 0-0.6% dei pazienti senza MEN 1 e, in una recente pubblicazione, Jensen ha sostenuto che il 37% dei pazienti affetti da MEN 1 sviluppa carcinoidi gastrici (5). Queste osservazioni suggeriscono che, nella ZES, l’ipergastrinemia non è sufficiente a provocare iperplasia delle cellule ECL, ma di solito altri fattori genetici sono necessari alla carcinoidogenesi. Sebbene non sia chiaro quali siano questi fattori, uno di essi potrebbe essere un fattore di crescita circolante, come quello che stimola l’iperplasia delle paratiroidi nei pazienti con MEN 1.
I tumori a cellule ECL nella MEN 1 sono spesso multicentrici, con assenza istologica di mitosi numerose; sono di dimensioni comprese tra pochi millimetri e 1.5 cm, e sono delimitati da mucosa e sottomucosa.
Non si conosce bene quale sia la percentuale di tumori endocrini dello stomaco associati a ZES/MEN 1 a comportamento invasivo; è noto che si possono osservare metastasi ai linfonodi locali nel 30% dei casi circa e metastasi a distanza in circa il 10% dei pazienti (2). Nonostante questo la sopravvivenza dei pazienti è generalmente eccellente anche in presenza di metastasi. Recentemente sono stati descritti da Bordi et al. carcinoidi gastrici di tipo 2 insolitamente aggressivi in due pazienti con MEN 1 (6).
{kind=link}
Tipo 3
I tumori endocrini dello stomaco di tipo 3 o sporadici si sviluppano generalmente a livello dell’antro gastrico, sono singoli, di dimesioni variabili tra 2 e 5 cm, non si associano ad ipergastrinemia e presentano generalmente un comportamento aggressivo con metastasi a distanza in circa il 50% dei casi (2). Nel 30-50% dei pazienti è presente la sindrome da carcinoide “atipica” caratterizzata da flush cutaneo di lunga durata, di tonalità purpurea associato a vasodilatazione ed edema del collo che fanno assumere al volto l’aspetto della “leonine face”. Altri sintomi associati sono: ipotensione, scialorrea, sudorazione, lacrimazione, rinorrea e reazione orticarioide pruriginosa.
TUMORE ENDOCRINO DEL DUODENO-DIGIUNO PROSSIMALE
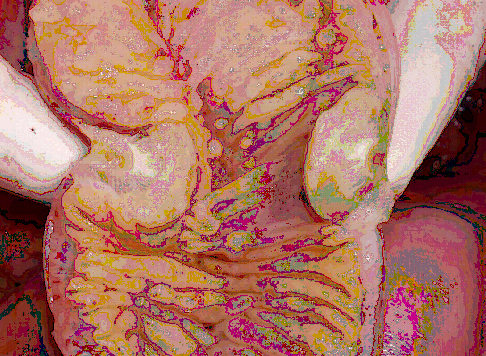
I tumori endocrini del duodeno (fig. 3) rappresentano circa il 6.3% dei tumori endocrini del tratto gastro-intestinale, presentano una maggiore incidenza nel sesso maschile e l’età media alla diagnosi è di circa 66 anni. Il riscontro è generalmente occasionale a seguito di una esofago-gastro-duodenoscopia eseguita per screening (15% dei casi) o per una sintomatologia aspecifica quale dispepsia (41% dei casi circa) o dolore addominale (48% dei pazienti). Più raramente la patologia si può manifestare con vomito, calo ponderale, emorragia digestiva o pancreatite acuta. La sindrome da carcinoide è rara e presente unicamente nei pazienti con malattia metastatica.
Generalmente i tumori endocrini del duodeno sono di piccole dimensioni e vengono trattati con la semplice escissione endoscopica. Nel caso in cui le dimensioni siano superiori al centimetro o la lesione si sia sviluppata in regione periampollare può essere necessario un approccio chirurgico tramite escissione transduodenale o cefalo-duodeno-pancreasectomia (7).
{kind=link}
TUMORI ENDOCRINI DEL DIGIUNO-ILEO-CIECO
I tumori endocrini dell’ileo (fig. 4) rappresentano la forma più frequente di neoplasia endocrina del tratto gastro-intestinale; l’età media alla diagnosi è di 66 anni e l’incidenza è lievemente maggiore nel sesso maschile (2). Essi sono composti da cellule enterocromaffini producenti serotonina e sostanza P. Tumori di dimensioni inferiori a 1 cm, non infiltranti la tonaca muscolare, hanno comportamento benigno, ma sono di solito riscontrati solo al tavolo autoptico, essendo totalemente asintomatici. Quelli diagnosticati in vivo sono, invece, di dimensioni superiori al centimetro, sono multipli nel 40% dei casi ed infiltrano a tutto spessore la tonaca muscolare. Per effetto della produzione tumorale di fattori di crescita, la parete muscolare infiltrata dal tumore appare ipertrofica e la proliferazione fibroblastica nella sottosierosa adiacente può indurre fenomeni di retrazione ed inginocchiamento dell’ansa intestinale coinvolta che possono produrre occlusione intestinale (8). (Prenota una visita gastroenterologica).
La patologia si può manifestare o con sintomi da massa neoplastica quale l’occlusione intestinale o con la classica sindrome da carcinoide. In quest’ultimo caso la produzione di amine biogene quali serotonina, idrossitriptamina, kallicreina e tachichinine determina lo sviluppo di una sintomatologia generalmente caratterizzata da flushing cutaneo (di colore da rosa chiaro a rosso, localizzato sul volto fino alla linea dei capezzoli, di breve durata e spesso scatenato da particolari alimenti), diarrea, broncospasmo e insufficienza tricuspidalica con fibrosi del cuore destro. Non di rado la diagnosi di tumore endocrino dell’ileo viene posta a seguito del riscontro occasionale, in corso di ecotomografia addominale, di metastasi epatiche multiple con lesione primitiva a sede ignota. In questi casi il ricorso a procedure diagnostiche quali la biopsia ecoguidata permette una valida caratterizzazione delle lesioni epatiche ed una corretta diagnosi.
{kind=link}
TUMORI ENDOCRINI DELL’APPENDICE
Comunemente conosciuti come carcinoidi appendicolari (fig. 5), rappresentano la grande maggioranza dei tumori endocrini di quest’organo. Essi sono in prevalenza di piccole dimensioni e costituiscono, di regola, un reperto occasionale in corso di appendicectomia per patologia infiammatoria. I carcinoidi appendicolari sono per lo più localizzati nella sottomucosa e nel contesto della tonaca muscolare, l’appendice rappresentando l’unico settore del tratto gastroenterico in cui l’infiltrazione della tonaca muscolare non comporta una diagnosi di malignità della neoplasia endocrina coinvolta.
I tumori endocrini dell’appendice rappresentano circa il 18% delle neoplasie endocrine del tratto gastro-intestinale, sono più frequenti nel sesso femminile e presentano un picco di incidenza tra i 15 ed i 19 anni per le donne e tra i 20 e i 29 anni per gli uomini (9). Sono generalmente lesioni benigne e solamente nel 5% circa dei casi è presente patologia metastatica, peraltro sempre per lesioni superiori a 2 cm di diametro. Clinicamente i tumori endocrini dell’appendice sono silenti e non si associano a sindrome da carcinoide.
{kind=link}
TUMORI ENDOCRINI DEL COLON-RETTO
I tumori endocrini del colon-retto sono prevalentemente localizzati nel retto (54%) essendo occasionali negli altri segmenti del grosso intestino (2). I tumori rettali sono di regola di piccole dimensioni (<1 cm), localizzati nella sottomucosa, sporgenti sulla superficie rettale come piccoli noduli polipoidi e presentano comportamento benigno dopo rimozione anche endoscopica. Carcinomi ben differenziati, identificati dall’infiltrazione della tonaca muscolare e/o dalla presenza di metastasi linfonodali o epatiche, rappresentano il 14% di tali neoplasie. Fattori di rischio per una evoluzione maligna sono un diametro superiore a 2 cm, presenza di angioinvasione, di più di 2 mitosi/10HPF e di aneuploidia. Clinicamente i tumori endocrini del retto sono generalmente asintomatici e diagnosticati in corso di esame endoscopico, raramente si presentano con rettorragia. (Prenota una visita chirurgica).
TUMORI ENDOCRINI DEL PANCREAS
INSULINOMA
L’insulinoma è il più comune tra i tumori funzionanti del pancreas, prevale nel sesso maschile, in età compresa tra 30 e 60 anni ed ha generalmente un comportamento benigno (10). Nel 70-80% dei casi il tumore si presenta come nodulo solitario, talvolta può avere multiple localizzazioni. Nella maggior parte dei casi si localizza nel pancreas, anche se insulinomi aberranti, sono stati ritrovati nel duodeno, nell’ileo, nel polmone e nella cervice uterina. E’ caratterizzato da una prevalente ed inappropriata secrezione di insulina che determina una sindrome caratterizzata da ipoglicemia (100%), sintomi neuroglicopenici (80-100%) e sintomi da stimolazione adrenergica (80-100%).
Si manifesta generalmente con sintomi subdoli quali fame, astenia, nausea, vomito, parestesie, neuropatie periferiche e visione offuscata. Tali sintomi sono in parte mascherati dalla tendenza di questi pazienti a compensare l’ipoglicemia con l’iperalimentazione; spesso quindi tali pazienti sono in soprappeso. I sintomi più drammatici dell’ipoglicemia quali le manifestazioni neuropsichiatriche oscillanti da moderati cambiamenti della personalità alla confusione mentale e al coma possono creare difficoltà nella diagnosi. (Prenota una visita endocrinologica).
GASTRINOMA
Il gastrinoma, tumore a prevalente secrezione di gastrina è responsabile della sindrome di Zollinger-Ellison (ZES) caratterizzata da malattia ulcerosa, ipersecrezione acida e/o diarrea (11). La sede prevalente del gastrinoma è il duodeno (76%) seguito dal pancreas (24%) (12). Sono stati descritti inoltre in letteratura gastrinomi nello stomaco, nell’intestino tenue e a livello linfonodale (13). Il gastrinoma è, nella maggior parte dei casi (70%), di piccole dimensioni e frequentemente multifocale, e per tale motivo, spesso non viene diagnosticato né con le comuni metodiche di imaging, né al tavolo operatorio. Le sedi più frequenti di metastatizzazione sono i linfonodi regionali (60%) e il fegato. Il gastrinoma determina frequentemente una malattia peptica (84-92%), per cui ogni lesione ulcerosa dovrebbe far nascere il sospetto di ZES. La diarrea può precedere, accompagnare o seguire l’ulcera; è permanente nel 25% dei casi, intermittente nel restante 75%. Il numero delle evacuazioni giornaliere può variare da 2-3 a più di 10, le feci sono chiare, acquose, untuose per la presenza di steatorrea. La diarrea è sempre associata ad ipergastrinemia e ipersecrezione acida, quindi facilmente distinguibile dalla diarrea della sindrome di Verner-Morrison da vipoma che costantemente è associata ad ipocloridria. (Prenota una visita gastroenterologica).
VIPOMA (SINDROME DI VERNER-MORRISON)
I termini vipoma, sindrome di Verner-Morrison (SVM), WDHA, rappresentano le denominazioni di un’affezione caratterizzata dalla presenza di un tumore endocrino a prevalente secrezione di VIP (Vaso Intestinal Polipeptide), caratterizzata da diarrea acquosa, ipokaliemia ed ipocloridria, in assenza di ipersecrezione acida e di ulcera peptica (14). Il sesso femminile sembra colpito con una frequenza tripla rispetto a quello maschile e la maggior parte delle osservazioni riguarda soggetti di età compresa fra i 17 e i 72 anni. Le neoplasie che si associano alla SVM sono per lo più di origine pancreatica, sebbene sempre più spesso vengano diagnosticati tumori di origine nervosa come ganglioneuromi, neuroblastomi e feocromocitomi che secernono VIP. La sindrome di Verner-Morrison è caratterizzata da crisi diarroiche coleriformi con notevole perdita di acqua ed elettroliti, con conseguente ipopotassiemia, dimagramento e disidratazione. Si associano inoltre intolleranza glucidica di tipo diabetico, ipercalcemia, ipomagnesemia, ipotensione e crisi di “flushing” cutaneo.
Circa la metà dei pazienti presenta una diarrea relativamente costante (10-15 scariche/die), mentre i rimanenti hanno episodi di abbondante diarrea alternati a periodi di diarrea meno severa. Negli stadi più avanzati, il paziente può evacuare, nelle 24 ore, fino a 10 litri di liquido ad elevato contenuto di elettroliti e bicarbonati. L’ipopotassiemia con valori di K+ sierico di solito inferiori a 3 mEq è responsabile dell’astenia muscolare, dei disturbi psichici, dell’insufficienza renale e dello scompenso cardiaco congestizio, elementi che con il passare del tempo diventano di primo piano nel quadro clinico. (Prenota una visita cardiologica). Il riscontro di diarrea acquosa refrattaria alla terapia e di ipopotassiemia, in assenza di malattie intestinali dimostrabili, deve far nascere il sospetto di SVM.
GLUCAGONOMA
Il glucagonoma è un tumore endocrino a prevalente secrezione di glucagone, è più frequente nel sesso femminile (55%) e l’età media alla diagnosi è di 65 anni (range 19-84); è localizzato nel 51% nella coda, nel 22% nella testa e nel 14% nel corpo del pancreas. Ha generalmente un comportamento maligno e si manifesta con una sindrome caratterizzata da eritema necrolitico migrante (90%), diabete mellito di varia gravità (87%) e dimagramento (96%) (15). A questi sintomi se ne associano altri quali trombosi venosa, diarrea, anemia normocromica normocitica e glossite. Spesso sono già presenti metastasi generalmente epatiche al momento della diagnosi. (Prenota un’ecografia epatica).
SOMATOSTATINOMA
Il somatostatinoma è un raro tumore a prevalente secrezione di somatostatina, più frequente nel sesso femminile ed è localizzato nel duodeno (57%) e nel pancreas (43%) (16). L’età media alla diagnosi è circa 53 anni, range 26-84. Le dimensioni variano da 2-3 cm per quelli duodenali a 5-6 cm per quelli pancreatici. Tra tutti i tumori neuroendocrini funzionanti del pancreas il somatostatinoma è sicuramente quello che ha una presentazione clinica più subdola poichè la sindrome è costituita da sintomi generici e di lieve entità. La classica sindrome è caratterizzata da litiasi della colecisti, diabete e diarrea con steatorrea. Il somatostatinoma duodenale può costituire un reperto accidentale nel corso di un esame endoscopico; si associa a sintomi e segni da ostruzione al deflusso bilio-pancreatico quando è localizzato nell’ampolla di Vater, oppure, talvolta con emorragia digestiva dovuta ad ulcerazione. (Prenota un’ecografia addominale). I somatostatinomi duodenali si associano frequentemente a neurofibromatosi di tipo I. Eccezionalmente il somatostatinoma si presenta nell’ambito della MEN 1.
TUMORI NON FUNZIONANTI DEL PANCREAS
Tali tumori vengono definiti non funzionanti in quanto non si associano sindromi cliniche da iperincreazione ormonale. Rappresentano circa il 40-60% di tutti i tumori endocrini del pancreas (17). Istologicamente sono costituiti da cellule a insulina (50%), PP (40%), glucagone (30%) e somatostatina (13%). Sono generalmente unifocali ad eccezione di quelli associati a MEN 1 che sono caratteristicamente multifocali. L’età di maggior incidenza è la quinta decade di vita (18).
La sintomatologia varia a seconda delle loro dimensioni, in fase iniziale sono generalmente asintomatici e quindi il loro riscontro è spesso occasionale durante accertamenti diagnostici per altri motivi, in fase più avanzata o quando presenti metastasi compaiono sintomi quali dolore epigastrico, vaghi dolori addominali, calo ponderale e ittero, determinati dall’effetto massa della neoplasia. Il loro silenzio clinico può essere dovuto alla produzione di forme peptidiche biologicamente inattive e/o alla secrezione di scarse quantità di peptidi, alla cosecrezione di peptidi inibitori come la somatostatina, e non ultimo alla down regulation dei recettori periferici. Nei pazienti affetti da tumore non funzionante del pancreas sono spesso presenti elevate concentrazioni sieriche e plasmatiche di PP, CgA, HCG, e neurotensina sostanze che peraltro non determinano sintomi particolari.
DIAGNOSI
MARCATORI ENDOCRINI GENERALI
Sono stati identificati alcuni altri componenti specifici per le cellule neuronali ed endocrine che possono essere utilizzati come marcatori tumorali: questi assumono un certo significato clinico in quanto possono essere utilizzati indipendentemente da peptidi e/o amine specifici prodotti dal tumore endocrino. Tali marcatori sono generalmente associati a granuli secretori o a piccole vescicole o sono presenti nel citosol delle cellule tumorali endocrine.
NSE (Neuro-specific-enolase)
E’ un isomero neuronospecifico dell’enzima glicolitico, ubiquitario, 2-phospho-Dglycerato hydrolase o enolase (19). Questo isomero è presente nei neuroni e nelle cellule endocrine e può essere utile come marcatore sierico per i tumori che derivano da queste cellule.
Cromogranina A
Le cromogranine costituiscono una famiglia di glicoproteine acidiche solubili in acqua, sono distribuite nei granuli secretori dei tessuti neuronali e neuroendocrini e si distinguono in cromogranina A, B (secretogranina I), C (secretogranina II) o pancreastatina e peptide NESP‑55 (neuroendocrine secretory). La più importante di tutte le cromogranine, in considerazione delle sue caratteristiche biologiche e della sua utilità clinica, è la Cromogranina A (CgA).
E’ stata descritta per la prima volta nel 1965 da Banks e Helle come proteina solubile isolata nei granuli cromaffini della midollare del surrene e nei neuroni del sistema nervoso simpatico (20). Tutte le cromogranine sono raccolte all’interno di granuli elettrondensi, contenenti anche altri ormoni peptidici, con i quali la CgA è cosecreta.
Studi successivi hanno dimostrato che la distribuzione di tale proteina era ben più complessa di quanto non si pensasse, essa infatti è contenuta nell’ipofisi (25%), nel pancreas (5%), nello stomaco (5%), nell’intestino (5%) e nelle rimanenti ghiandole endocrine (1%). In piccola quantità è stata isolata anche dalle cellule neuroendocrine della ghiandola mammaria, del polmone e della prostata, nelle ghiandole sottomandibolari, nella milza, nei linfonodi nel timo e nel fegato. La distribuzione tissutale delle altre cromogranine non è stata ancora del tutto definita, ma sembra che esse siano presenti in molti tessuti.
Il ruolo fisiologico di queste molecole non è chiaro; probabilmente esse intervengono nella regolazione della funzione dei granuli secretori; inoltre potrebbero essere precursori di peptidi biologicamente attivi (pro-ormoni).
E’ stato dimostrato che le cromogranine sono marcatori immunocitochimici dei tessuti neuroendocrini e quindi hanno valore diagnostico nei tumori da questi derivano. In particolare si è visto che la CgA, largamente distribuita nelle cellule endocrine e neuroendocrine che producono peptidi, è un utile marcatore istologico di tumore endocrino benigno e maligno di varia origine.
Recentemente è stato messo a punto il dosaggio immunologico della CgA plasmatica, e dai dati della letteratura questa rappresenta un utile marcatore bioumorale di neoplasia endocrina. Livelli elevati di CgA sono stati dimostrati anche in pazienti affetti da neoplasia prostatica e sono espressione della differenziazione neuroendocrina di una parte delle cellule che costituiscono la ghiandola. La CgA potrebbe avere un ruolo nel follow-up dei pazienti con carcinoma prostatico con PSA negativo.
Esistono altre condizioni patologiche in cui la CgA potrebbe essere aumentata quali per esempio l’insufficienza renale, la gastrite cronica atrofica e l’insufficienza respiratoria che vanno quindi attentamente valutate. (Prenota una visita endocrinologica).
MARCATORI ENDOCRINI SPECIFICI
La diagnosi di tumore endocrino si basa, oltre che su una attenta valutazione della storia clinica, sull’individuazione dei marcatori bioumorali responsabili delle varie sindromi cliniche e sulla localizzazione del tumore e delle sue eventuali metastasi. A tale scopo può essere sufficiente il loro valore basale, ma più spesso si rende necessaria l’esecuzione di test di stimolo. I tests funzionali sono molto importanti poiché l’elevato livello circolante di un determinato ormone gastrointestinale non è necessariamente espressione della presenza di tumore. Esistono infatti situazioni patologiche, quali ad esempio la gastrite cronica atrofica o l’insufficienza renale cronica, in cui i livelli di gastrinemia si dimostrano marcatamente elevati con valori simili a quelli riscontrabili nei gastrinomi. Elevati livelli di un ormone gastrointestinale possono essere anche conseguenti a proliferazione di tipo non neoplastico delle cellule endocrine come ad esempio l’iperplasia. L’applicazione di tests evocativi e/o di soppressione diventa quindi particolarmente utile nella diagnosi differenziale tra iperplasia e tumore. Infatti le cellule del sistema endocrino diffuso rispondono appropriatamente alla regolazione degli stimoli esogeni, viceversa quelle tumorali rispondono in maniera del tutto autonoma ed in maniera paradossa, consentendo così la differenziazione diagnostica.
Nell’insulinoma, l’ipoglicemia stimolata dall’esercizio fisico o dal digiuno con le relative manifestazioni dovute alla diminuita concentrazione di glucosio a livello cerebrale e all’eccesso di catecolamine, liberate nel tentativo di sopperire all’abbassamento della glicemia, sono i sintomi cardine provocati dall’eccessiva secrezione di insulina. Per la diagnosi di insulinoma di fondamentale importanza risulta essere il test al digiuno che è positivo quando la glicemia diminuisce a 40 mg/dl dopo 24-72 ore di digiuno.
Nel gastrinoma la diagnosi biochimica di sindrome di Zollinger-Ellison si basa sul riscontro di elevati valori di gastrinemia basale (> 1000 pg/dl) associati ad un basso pH gastrico (<2). In presenza di una gastrinemia basale compresa tra 100 e 1000 pg/dl con pH gastrico < 2, per la diagnosi di gastrinoma è indicata l’esecuzione del test alla secretina che risulta positivo se vi è un aumento della gastrina plasmatica > di 200 pg/ml dopo 2-5 minuti dall’infusione di Secretina (Fig. 6). Perché i dosaggi di gastrina siano attendibili è necessario che i prelievi vengano eseguiti dopo almeno sette giorni di sospensione di farmaci inibitori della pompa protonica, oppure dopo 24 ore dall’assunzione di H2 antagonisti.
Per quanto riguarda il glucagonoma, il Vipoma ed il somatostatinoma non esistono test provocativi attendibili ed il dosaggio dei rispettivi ormoni in circolo non sempre è attendibile.
Per quanto riguarda la diagnosi di tumore endocrino del tratto gastro-intestinale i principali mediatori della sindrome da carcinoide, sia nella forma tipica che atipica sono serotonina, Kallicreina, tachichinine, prostaglandine, 5-idrossitriptofano e l’istamina quest’ultima soprattutto nella sindrome atipica. Il dosaggio del metabolica urinario della serotonina (acido 5-idrossi-indolo-acetico: 5-HIAA) rappresenta tuttora il cardine della diagnosi bioumorale della sindrome da carcinoide. In particolare la sensibilità diagnostica del 5-HIAA urinario è secondo molti autori compresa tra il 65-75% con una specificità pari al 90-100% (21). Valori di riferimento sono compresi tra 2 e 8 mg/24 h, mentre valori > 20 mg/24 h sono considerati “cut-off” al di sopra dei quali la diagnosi è certa. Valori lievemente superiori alla norma pongono il problema dei falsi positivi provocati generalmente dall’assunzione di cibi ricchi in serotonina come banane, avocados, kiwi, cacao, arachidi, oppure farmaci come salicilati, L-dopa e guaiacolati e betabloccanti. Errori nelle procedure di raccolta e conservazione delle urine che va fatta in appositi contenitori oscurati, tenuti lontano da fonti di calore e/o luce, possono anch’essi dare luogo a falsi risultati.
{kind=link}
DIAGNOSI STRUMENTALE
Le tecniche di imaging svolgono un ruolo centrale per la diagnosi e la corretta stadiazione delle neoplasie endocrine del tratto gastro-entero-pancreatico.
Attualmente sono disponibili numerose metodiche d’indagine invasive e non invasive:
1 Ultrasonografia con e senza mezzo di contrasto;
2 Ultrasonografia endoscopica (EUS)
3 Tomografia Assiale Computerizzata (TC);
4 Risonanza Magnetica Nucleare (RM);
5 PET;
6 Scintigrafia con Octreoscan.
Ultrasonografia
L’ecografia addominale occupa attualmente un posto di primo piano fra le metodiche strumentali utilizzate nella diagnostica e nella stadiazione dei tumori endocrini del tratto GEP. (Prenota un’ecografia addominale).
Numerosi studi hanno valutato la sensibilità e la specificità dell’ultrasonografia nella diagnosi di tumore endocrino del pancreas. In particolare diversi autori hanno riportato una sensibilità variabile tra il 25 ed il 70% per l’insulinoma e del 30% per il gastronoma (22-24). Nel 1991 uno studio prospettico ha permesso di valutare che la sensibilità di tale metodica nella localizzazione dei gastrinomi è correlata alle dimensioni della lesione; infatti nessun gastrinoma extraepatico di dimensioni inferiori ad 1 cm è stato identificato con l’ecografia, mentre la sensibilità cresce al 15% nei tumori con dimensioni da 1 a 3 cm e risulta del 100% nei tumori con dimensioni superiori a 3 cm (23).
I dati in letteratura sull’utilizzo dell’ecotomografia nella diagnosi dei tumori GEP più rari sono limitati. Comunque, considerando che le dimensioni di tali tumori sono in genere maggiori, al momento della diagnosi, di quelle dei gastrinomi e degli insulinomi, l’ecografia può essere ugualmente utilizzata con una sensibilità sufficientemente alta.
Ultrasonografia endoscopica (EUS)
Una recente applicazione dell’ultrasonografia riguarda la sua associazione con l’endoscopia digestiva che consente di diagnosticare tumori pancreatici di dimensioni di 2-3 mm (25). Infatti, in uno studio multicentrico condotto su soggetti affetti da tumori endocrini del pancreas, non evidenziati con TC ed ecografia, l’EUS ha localizzato correttamente la neoformazione nell’82% dei casi con una riduzione della sensibilità al 60% per la localizzazione corpo-caudale. Un recente studio prospettico ha mostrato una sensibilità del 93% ed una specificità del 95% nelle localizzazione delle lesioni intrapancreatiche (26).
Tale metodica è peraltro dotata di una buona accuratezza diagnostica anche per tumori a sede extraepatica (90% per la lesione primitiva) e per le loro metastasi (75%) come si può desumere da uno studio su un gruppo di soggetti con neoplasie endocrine localizzate allo stomaco, duodeno e retto (27).
Tomografia Assiale Computerizzata
La maggioranza delle neoformazioni endocrine del pancreas sono isodense alla TC e possono non essere visualizzate senza l’utilizzo del mezzo di contrasto mentre dopo iniezione di mezzo di contrasto assumono il comportamento delle lesioni ipervascolarizzate.
Recentemente la TC-spirale (hCT), evoluzione tecnica della tomografia computerizzata tradizionale, caratterizzata dalla possibilità di avere immagini sia della fase arteriosa che di quella venosa dopo singola iniezione di mezzo di contrasto, sembra costituire un significativo vantaggio nella localizzazione dei tumori endocrini. Numerosi Autori hanno riportato, per tale metodica, una sensibilità variabile tra l’82 ed il 92% (24;28).
Risonanza Magnetica Nucleare
Negli ultimi anni la Risonanza Magnetica Nucleare (RMN) ha assunto un ruolo sempre più importante nella diagnosi e nella stadiazione dei tumori endocrini del pancreas e delle metastasi epatiche (29). Una rivalutazione delle più recenti pubblicazioni accorda alla RMN una sensibilità del 91% nella diagnosi delle lesioni pancreatiche; mentre tale metodica è considerata la tecnica più sensibile per dimostrare la presenza di metastasi epatiche ed ossee (30).
PET
Negli ultimi anni, numerosi studi hanno cercato di valutare la sensibilità e la specificità della PET nella diagnosi e nella stadiazione dei tumori endocrini del tratto gastro-entero-pancreatico (31). In particolare si è evidenziato come il 18F-FDG, generalmente utilizzato nello studio delle patologie neoplastiche, abbia un valore limitato nella diagnostica dei tumori endocrini poiché questi generalmente non presentano una sufficiente captazione dell’FDG. Più di recente, grazie alla disponibilità di traccianti chimici specifici per le cellule endocrine, come il 18F-FDOPA, la PET ha assunto una maggiore importanza nella diagnosi e stadiazione delle neoplasie endocrine (32). (Prenota una visita endocrinologica).
Di particolare interesse, a tal proposito, risulta un recente lavoro di Becherer et al che ha raffrontato tra loro alcune metodiche quali, la PET con 18F-FDOPA, la scintigrafia con Octreoscan e la TC nella stadiazione dei tumori endocrini. Tale studio ha evidenziato come la PET permetta di ottenere una migliore localizzazione della lesione neoplastica rispetto alla scintigrafia con Octreoscan con una sensibilità maggiore rispetto alla TC nella identificazione di metastasi ossee (32).
Scintigrafia con Octreoscan
I recettori per la somatostatina sono presenti in alcuni tumori che originano da tessuti che fisiologicamente presentano tali recettori sulle membrane cellulari. Un gran numero di recettori ad alta affinità sono stati trovati nella maggioranza dei tumori primitivi e metastatici del Sistema Neuroendocrino Diffuso. Questo dato ha spinto i ricercatori a cercare di visualizzare tumori con recettori positivi in vivo usando un analogo marcato con iodio radioattivo.
Attualmente, data la semplicità di preparazione, la buona disponibilità e la accettabile interferenza nella visualizzazione di lesioni addominali, il radiofarmaco di scelta per la scintigrafia dei recettori per la SST è lo [“In-DTPA-D-Phel]-octreotide, denominato OCTREOSCAN.
I tumori endocrini gastroenteropancreatici (GEP), siano essi funzionanti o non funzionanti, presentano nella maggior parte dei casi, recettori per la SST e pertanto fissano l’Octreoscan. Secondo la letteratura, l’esame con Octreoscan può consentire di localizzare tumori di diametro inferiore ai 2 cm. e di evidenziare con elevata sensibilità anche localizzazioni extrapancreatiche ed extraepatiche, di difficile rilievo con altre metodiche di imaging (33).
Gli adenocarcinomi del pancreas esocrino non vengono visualizzati dal radiofarmaco, mentre gli insulinomi fissano in circa il 50% dei casi l’indicatore radioattivo. In uno studio di Krenning non è stata ottenuta alcuna immagine nei 24 pazienti portatori di carcinoma pancreatico primitivo, mentre il 61% degli insulinomi è stato visualizzato. Questo dato di relativa bassa positività di fissazione per quanto riguarda gli insulinomi fa ipotizzare la presenza di un sottotipo di recettori per la SST sulla loro membrana cellulare con minore affinità per il radiocomposto. In uno studio di confronto tra la Tomografia a fotone singolo (SPECT) con Octreoscan, la Risonanza Magnetica e la Tomografia Computerizzata effettuato su 17 pazienti con tumori GEP, si è dimostrato che la metodica scintigrafica possiede la maggiore accuratezza, soprattutto in quei casi in cui l’esame clinico e le indagini di laboratorio non risultano dirimenti.
TERAPIA
Chemioterapia
E’ stata per lungo tempo impiegata nel trattamento dei tumori endocrini metastatici, attualmente il suo impiego è limitato alle forme altamente indifferenziate (34-36).
I farmaci più frequentemente utilizzati sono la streptozotocina (STZ), il 5-fluorouracile (5-FU), la doxorubicina (DOX), l’etoposide (ETP) e il cisplatino (DPP). L’associazione più vantaggiosa sembra essere: STZ 0,5 g/m2 + 5-FU 400 mg/m2, per 5 giorni, ogni 6 settimane. Con tale associazione si è ottenuta una risposta obiettiva nel 50-60% dei pazienti con durata della remissione fino a 24 mesi ed un tempo medio di sopravvivenza di circa 26 mesi. Risultati simili si sono ottenuti con un’altra associazione: STZ 0,5 g/m2 + DOX 40 mg/m2, per 5 giorni, ogni 6 settimane. Il DPP e l’ETP vengono impiegati in combinazione, nelle forme scarsamente differenziate, con il seguente schema: DPP 45 mg/m2 per 2 giorni ed ETP 130 mg/m2 per i successivi 3 giorni, ogni 4 settimane. La percentuale di risposta è del 60-70%, ma la durata della remissione è breve (6-8 mesi). L’impiego dei chernioterapici è tuttavia gravato da notevoli effetti collaterali che ne limitano l’uso quali: nausea, vomito, alterazioni della crasi ematica e, soprattutto, compromissione della funzione renale.
E’ importante sottolineare che i tumori funzionanti sono più responsivi alla chemioterapia dei non funzionanti e che nell’ambito dei funzionanti i carcinoidi sono meno chemosensibili dei tumori del pancreas.
Embolizzazione e chemioembolizzazione
Un utile approccio terapeutico, in pazienti inoperabili e/o inadatti alla chemioterapia, è rappresentato dalla embolizzazione dell’arteria epatica, che determina una riduzione di volume della massa tumorale per necrosi ischemica. Tale metodica è ripetibile e può essere eseguita in combinazione con una chemioterapia sia sistemica, che locale; in quest’ultimo caso il chemioterapico, es. STZ, viene miscelato con l’agente embolizzante (gelfoam).
Dai dati della letteratura così come nella nostra esperienza, la chemioembolizzazione si è dimostrata efficace sia nel controllo dei sintomi in quanto riduce la secrezione ormonale, sia nel controllo della proliferazione del tumore. Generalmente, tali procedure sono ben tollerate dal paziente e gli effetti collaterali più frequenti, nausea dolore addominale, febbre, definiti come sindrome post-chemioembolizzazione, sono di lieve entità e transitori (37;38).
Interferone
L’interferone è stato utilizzato nella terapia dei tumori endocrini per la sua capacità di inibire la secrezione ormonale e di controllare la crescita tumorale mediante il blocco del ciclo cellulare in G0, G1, l’induzione della 2’-5’ A sintetasi, la modulazione del sistema immunitario e l’inibizione dell’angiogenesi. Esso riduce l’espressione dell’mRNA per i vari peptidi e determina apoptosi delle cellule tumorali. Il dosaggio più comunemente utilizzato è di 5-6 MU/3-5 volte alla settimana con il quale si ottiene una risposta obiettiva e biochimica nel 50% e tumorale nel 12% dei pazienti, con una durata media di risposta di circa 20 mesi. Recenti studi hanno dimostrato, che tali percentuali di risposta migliorano nettamente quando si associano all’interferone gli analoghi della somatostatina (risposta obiettiva nel 68.5% e tumorale nel 50% dei pazienti). Gli effetti collaterali più frequenti sono rappresentati dalla sindrome simil influenzale e dalla mielodepresisone che generalmente sono di lieve entità (39;40).
Analoghi della somatostatina
L’evidenza che la somatostatina e soprattutto i suoi analoghi abbiano una azione inibente sulla secrezione di molti peptidi e su varie funzioni digestive oltre ad un probabile effetto antiproliferativo ne hanno suggerito l’impiego nella terapia dei tumori neuroendocrini. Il razionale per l’uso di tali farmaci è la presenza, dimostrata con l’autoradiografia e la scintigrafia con octreoscan, di recettori per la somatostatina nell’80-90% dei tumori neuroendocrini. Infatti sono stati identificati 5 diversi recettori per la somatostatina codificati da geni presenti in cromosomi distinti e con diversa funzione nei vari organi. I tipi 1, 2 e 5 controllano la proliferazione cellulare, i recettori 1 e il 2 mediante l’attivazione della tirosina fosfatasi, il 5 con meccanismo che coinvolge probabilmente variazioni della mobilizzazione del calcio intracellulare. Tutti i tipi recettoriali legano la somatostatina; l’octreotide con alta affinità i sottotipi recettoriali 2 e 5 e con moderata affinità il sottotipo 3 ma non lega i sottotipi 1 e 4. Infine gli stessi analoghi ad alte dosi, si legano ai recettori 3 che mediano l’apoptosi.
L’impiego della somatostatina nativa nel trattamento a lungo termine è limitato dalla sua breve emivita che rende necessaria l’ infusione continua. La sintesi nel 1982, di octreotide acetato, derivato octapeptidico a più lunga emivita (circa 8 ore) ha notevolmente migliorato la qualità di vita dei pazienti affetti da tali tumori.
L’octreotide è ampiamente utilizzato nella pratica clinica, nel controllo della sindrome da carcinoide, e nel trattamento delle varie sindromi da iperincrezione ormonale (41;42). Inoltre viene utilizzato come farmaco d’emergenza in corso di crisi acuta scatenata da interventi e/o manovre chirurgiche. Gli analoghi della somatostatina vengono altresì impiegati nel trattamento dei tumori neuroendocrini non funzionanti per il loro effetto antiproliferativo mediato dai recettori della somatostatina. Altri meccanismi coinvolti sembrano essere l’inibizione dell’angiogenesi, del release degli ormoni trofici e la modulazione dell’attività immunologica.
Alcuni pazienti in trattamento cronico con tale farmaco richiedono un progressivo incremento delle dosi (tachifilassi) dovuto ad una «down regulation» recettoriale e ad aumento di volume della massa tumorale.
Recentemente sono stati ottenuti nuovi analoghi a più lunga durata d’azione in grado di fornire, mediante un meccanismo di lento rilascio, concentrazioni plasmatiche stabili ed efficaci per 14-28 giorni. Gli analoghi long-acting attualmente disponibili sono octreotide acetato LAR e lanreotide. Octreotide acetato LAR è costituito da microsfere di un polimero biodegradabile, ed è disponibile a dosaggio di 10, 20 e 30 mg somministrabile mediante iniezione intramuscolare profonda ogni 28 giorni. Lanreotide è anch’esso incorporato in microsfere biodegradabili che consentono un lento rilascio mantenendo livelli efficaci del farmaco per 10-14 giorni al dosaggio di 30 mg, e per 28 giorni al dosaggio di 60 mg. Il vantaggio che tali analoghi offrono, rispetto all’octreotide s.c., è legato al miglioramento della compliance del paziente con notevole impatto sulla qualità di vita.
Gli studi disponibili in letteratura riportano una efficacia pari a quella di octreotide s.c. sia in termini di controllo dei sintomi che di stabilizzazione del tumore ma con il grande vantaggio di somministrazioni bisettimanali o mensili. Noi abbiamo valutato l’efficacia di Octreotide LAR in 16 pazienti affetti da diversi tipi di tumore neuroendocrino gastrointestinale dimostrando un buon controllo sui sintomi in tutti i pazienti ed una stabilizzazione della malattia nell’87,5% per un periodo di 10.7 mesi (43).
Recentemente abbiamo osservato in tre pazienti affetti da carcinoidosi gastrica associata a sindrome MEN 1 la scomparsa delle lesioni dopo un anno di trattamento con analoghi long-acting della somatostatina (44).
Pochi studi hanno chiarito il potenziale valore della terapia dei tumori neuroendocrini metastatici con analoghi ad alte dosi (45;46). Studi riportati da Eriksson e Faiss hanno dimostrato che alte dosi di analoghi long-acting comportano una risposta biochimica, con controllo dei sintomi e stabilizzazione della massa tumorale nella maggior parte dei pazienti. Di particolare interesse è stata l’osservazione che analoghi long-acting ad alte dosi aumentano il numero delle cellule apoptotiche dopo 6-8 mesi di trattamento. Ciò suggerisce che gli analoghi somministrati a tali dosaggi possano essere utilizzate quando non vi è risposta alle dosi standard .
Gli effetti collaterali degli analoghi della somatostatina sono generalmente modesti, spesso dovuti all’adattamento del sistema gastroenterico al farmaco. Tra questi i più frequenti sono: nausea, flatulenza, dolori addominali crampiformi, diarrea e, dolore nella sede dell’ iniezione. Si possono inoltre verificare alterazioni del metabolismo glucidico nel senso di una ridotta tolleranza per inibizione della secrezione di insulina da parte di octreotide. Per contro, lanreotide e octreotide LAR determinano una riduzione della glicemia nei pazienti con diabete mellito per una maggior inibizione della secrezione di GH e glucagone rispetto all’insulina. Nel trattamento a lungo termine si può sviluppare una colelitiasi, generalmente asintomatica, che sembra interessare, secondo alcuni autori, il 20-50 % dei soggetti.
Terapia radiometabolica
In recenti studi alcuni ricercatori hanno cercato di sviluppare un anologo della somatostatina ad alta affinità recettoriale che possa essere legato a un isotopo beta-emittente. Il complesso formato da radioisotopo e anologo si lega alle cellule che contengono recettori per la somatostatina e le distrugge. La nuova generazione di radioisotopi è costituita dal chelante DOTA associato a 90Y (DOTATOC) o a 177Lu (DOTATATE). Molti lavori sottolineano l’utilità di tale terapia nei tumori endocrini, in particolare Kwekkeboom et al. hanno studiato l’effetto della terapia con 117Lu nei pazienti con tumori gastroenteropancreatici, ottenendo una risposta parziale nel 20-63% dei casi e una malattia stabile nel 12-42% dei soggetti. L’effetto della terapia è maggiore nei pazienti con lesioni di dimensioni limitate ed è quindi preferibile considerare questo trattamento nelle fasi iniziali della patologia. Fino ad oggi non sono stati riportati seri effetti collaterali (47).
Prof. Paola Tomassetti
Bibliografia
1. Solcia E, Kloppel G, Sobin LH. Histological typing of endocrine tumors. And ed. Heidelberg: 2000.
2. Modlin IM, Lye KD, Kidd M. A 5-decade analysis of 13,715 carcinoid tumors. Cancer 2003; 97(4): 934-59.
3. Rindi G, Luinetti O, Cornaggia M, Capella C, Solcia E. Three subtypes of gastric argyrophil carcinoid and the gastric neuroendocrine carcinoma: a clinicopathologic study. Gastroenterology 1993; 104(4): 994-1006.
4. Rindi G, Bordi C, Rappel S, La Rosa S, Stolte M, Solcia E. Gastric carcinoids and neuroendocrine carcinomas: pathogenesis, pathology, and behavior. World J Surg 1996; 20(2): 168-72.
5. Jensen RT. Management of the Zollinger-Ellison syndrome in patients with multiple endocrine neoplasia type 1. J Intern Med 1998; 243(6): 477-88.
6. Bordi C, Falchetti A, Azzoni C, D’Adda T, Canavese G, Guariglia A, Santini D, Tomassetti P, Brandi ML. Aggressive forms of gastric neuroendocrine tumors in multiple endocrine neoplasia type I. Am J Surg Pathol 1997; 21(9): 1075-82.
7. Zyromski NJ, Kendrick ML, Nagorney DM, Grant CS, Donohue JH, Farnell MB, Thompson GB, Farley DR, Sarr MG. Duodenal carcinoid tumors: how aggressive should we be? J Gastrointest Surg 2001; 5(6): 588-93.
8. Bordi C. Aspetti anatomopatologici dei tumori endocrini gastroenteropancreatici. In: Rosato L, editor. I tumori neuroendocrini. CLUB delle U.E.C.; 2003. p. 48-54.
9. Hemminki K, Li X. Incidence trends and risk factors of carcinoid tumors: a nationwide epidemiologic study from Sweden. Cancer 2001; 92(8): 2204-10.
10. Wilder R, Allan R, Power M, Robertsson H. Carcinoma of the island of the pancreas, hiperinsulinemia and hypoglycemia. JAMA 1927; 89: 348-55.
11. Zollinger RM, Ellison EH. Primary peptic ulcerations of the jejunum associated with islet cell tumors of the pancreas. 1955. CA Cancer J Clin 1989; 39(4): 231-47.
12. Norton JA, Jensen RT. Current surgical management of Zollinger-Ellison syndrome (ZES) in patients without multiple endocrine neoplasia-type 1 (MEN1). Surg Oncol 2003; 12(2): 145-51.
13. Norton JA, Alexander HR, Fraker DL, Venzon DJ, Gibril F, Jensen RT. Possible primary lymph node gastrinoma: occurrence, natural history, and predictive factors: a prospective study. Ann Surg 2003; 237(5): 650-7.
14. VERNER JV, MORRISON AB. Islet cell tumor and a syndrome of refractory watery diarrhea and hypokalemia. Am J Med 1958; 25(3): 374-80.
15. Mallinson CN, Bloom SR, Warin AP, Salmon PR, Cox B. A glucagonoma syndrome. Lancet 1974; 2(7871): 1-5.
16. Vinik AI, Strodel WE, Eckhauser FE, Moattari AR, Lloyd R. Somatostatinomas, PPomas, neurotensinomas. Semin Oncol 1987; 14(3): 263-81.
17. Tomassetti P, Campana D, Piscitelli L, Casadei R, Santini D, Nori F, Morselli-Labate AM, Pezzilli R, Corinaldesi R. Endocrine pancreatic tumors: factors correlated with survival. Ann Oncol 2005; 16(11): 1806-10.
18. Gullo L, Migliori M, Falconi M, Pederzoli P, Bettini R, Casadei R, Delle FG, Corleto VD, Ceccarelli C, Santini D, Tomassetti P. Nonfunctioning pancreatic endocrine tumors: a multicenter clinical study. Am J Gastroenterol 2003; 98(11): 2435-9.
19. Schmechel D, Marangos PJ, Brightman M. Neurone-specific enolase is a molecular marker for peripheral and central neuroendocrine cells. Nature 1978; 276(5690): 834-6.
20. Winkler H, Fischer-Colbrie R. The chromogranins A and B: the first 25 years and future perspectives. Neuroscience 1992; 49(3): 497-528.
21. Feldman JM, O’Dorisio TM. Role of neuropeptides and serotonin in the diagnosis of carcinoid tumors. Am J Med 1986; 81(6B): 41-8.
22. King CM, Reznek RH, Dacie JE, Wass JA. Imaging islet cell tumours. Clin Radiol 1994; 49(5): 295-303.
23. London JF, Shawker TH, Doppman JL, Frucht HH, Vinayek R, Stark HA, Miller LS, Miller DL, Norton JA, Jensen RT. Zollinger-Ellison syndrome: prospective assessment of abdominal US in the localization of gastrinomas. Radiology 1991; 178(3): 763-7.
24. Thoeni RF, Mueller-Lisse UG, Chan R, Do NK, Shyn PB. Detection of small, functional islet cell tumors in the pancreas: selection of MR imaging sequences for optimal sensitivity. Radiology 2000; 214(2): 483-90.
25. Glover JR, Shorvon PJ, Lees WR. Endoscopic ultrasound for localisation of islet cell tumours. Gut 1992; 33(1): 108-10.
26. Anderson MA, Carpenter S, Thompson NW, Nostrant TT, Elta GH, Scheiman JM. Endoscopic ultrasound is highly accurate and directs management in patients with neuroendocrine tumors of the pancreas. Am J Gastroenterol 2000; 95(9): 2271-7.
27. Yoshikane H, Tsukamoto Y, Niwa Y, Goto H, Hase S, Mizutani K, Nakamura T. Carcinoid tumors of the gastrointestinal tract: evaluation with endoscopic ultrasonography. Gastrointest Endosc 1993; 39(3): 375-83.
28. Van HL, Gryspeerdt S, Marchal G, Baert AL, Mertens L. Helical CT for the preoperative localization of islet cell tumors of the pancreas: value of arterial and parenchymal phase images. AJR Am J Roentgenol 1995; 165(6): 1437-9.
29. Ichikawa T, Peterson MS, Federle MP, Baron RL, Haradome H, Kawamori Y, Nawano S, Araki T. Islet cell tumor of the pancreas: biphasic CT versus MR imaging in tumor detection. Radiology 2000; 216(1): 163-71.
30. Debray MP, Geoffroy O, Laissy JP, Lebtahi R, Silbermann-Hoffman O, Henry-Feugeas MC, Cadiot G, Mignon M, Schouman-Claeys E. Imaging appearances of metastases from neuroendocrine tumours of the pancreas. Br J Radiol 2001; 74(887): 1065-70.
31. Eriksson B, Bergstrom M, Orlefors H, Sundin A, Oberg K, Langstrom B. Use of PET in neuroendocrine tumors. In vivo applications and in vitro studies. Q J Nucl Med 2000; 44(1): 68-76.
32. Becherer A, Szabo M, Karanikas G, Wunderbaldinger P, Angelberger P, Raderer M, Kurtaran A, Dudczak R, Kletter K. Imaging of advanced neuroendocrine tumors with (18)F-FDOPA PET. J Nucl Med 2004; 45(7): 1161-7.
33. Gibril F, Reynolds JC, Doppman JL, Chen CC, Venzon DJ, Termanini B, Weber HC, Stewart CA, Jensen RT. Somatostatin receptor scintigraphy: its sensitivity compared with that of other imaging methods in detecting primary and metastatic gastrinomas. A prospective study. Ann Intern Med 1996; 125(1): 26-34.
34. Bajetta E, Ferrari L, Procopio G, Catena L, Ferrario E, Martinetti A, Di Bartolomeo M, Buzzoni R, Celio L, Vitali M, Beretta E, Seregni E, Bombardieri E. Efficacy of a chemotherapy combination for the treatment of metastatic neuroendocrine tumours. Ann Oncol 2002; 13(4): 614-21.
35. Kaltsas GA, Mukherjee JJ, Isidori A, Kola B, Plowman PN, Monson JP, Grossman AB, Besser GM. Treatment of advanced neuroendocrine tumours using combination chemotherapy with lomustine and 5-fluorouracil. Clin Endocrinol (Oxf) 2002; 57(2): 169-83.
36. Oberg K. Advances in chemotherapy and biotherapy of endocrine tumors. Curr Opin Oncol 1998; 10(1): 58-65.
37. Dominguez S, Denys A, Madeira I, Hammel P, Vilgrain V, Menu Y, Bernades P, Ruszniewski P. Hepatic arterial chemoembolization with streptozotocin in patients with metastatic digestive endocrine tumours. Eur J Gastroenterol Hepatol 2000; 12(2): 151-7.
38. Kress O, Wagner HJ, Wied M, Klose KJ, Arnold R, Alfke H. Transarterial chemoembolization of advanced liver metastases of neuroendocrine tumors–a retrospective single-center analysis. Digestion 2003; 68(2-3): 94-101.
39. Fjallskog ML, Sundin A, Westlin JE, Oberg K, Janson ET, Eriksson B. Treatment of malignant endocrine pancreatic tumors with a combination of alpha-interferon and somatostatin analogs. Med Oncol 2002; 19(1): 35-42.
40. Eriksson B., Oberg K. Interferon Therapy of malignant endocrine pancreatic tumors. Mignon M., Jensen R.T., editors. Endocrine tumors of the pancreas: Recent advances in research and management. Karger[23], 451-460. 1995. Frontiers of Gastrointestinal Research. Modlin I.M., Rozen P., and Scarpignato C. Ref Type: Serial (Book,Monograph)
41. de Herder WW, Krenning EP, van Eijck CH, Lamberts SW. Considerations concerning a tailored, individualized therapeutic management of patients with (neuro)endocrine tumours of the gastrointestinal tract and pancreas. Endocr Relat Cancer 2004; 11(1): 19-34.
42. Arnold R, Simon B, Wied M. Treatment of neuroendocrine GEP tumours with somatostatin analogues: a review. Digestion 2000; 62 Suppl 1: 84-91.
43. Tomassetti P, Migliori M, Corinaldesi R, Gullo L. Treatment of gastroenteropancreatic neuroendocrine tumours with octreotide LAR. Aliment Pharmacol Ther 2000; 14(5): 557-60.
44. Tomassetti P, Migliori M, Caletti GC, Fusaroli P, Corinaldesi R, Gullo L. Treatment of type II gastric carcinoid tumors with somatostatin analogues. N Engl J Med 2000; 343(8): 551-4.
45. Faiss S, Rath U, Mansmann U, Caird D, Clemens N, Riecken EO, Wiedenmann B. Ultra-high-dose lanreotide treatment in patients with metastatic neuroendocrine gastroenteropancreatic tumors. Digestion 1999; 60(5): 469-76.
46. Eriksson B, Renstrup J, Imam H, Oberg K. High-dose treatment with lanreotide of patients with advanced neuroendocrine gastrointestinal tumors: clinical and biological effects. Ann Oncol 1997; 8(10): 1041-4.
47. Kwekkeboom DJ, Teunissen JJ, Bakker WH, Kooij PP, de Herder WW, Feelders RA, van Eijck CH, Esser JP, Kam BL, Krenning EP. Radiolabeled somatostatin analog [177Lu-DOTA0,Tyr3]octreotate in patients with endocrine gastroenteropancreatic tumors. J Clin Oncol 2005; 23(12): 2754-62.
SET
2011