CUSHING DA SECREZIONE ECTOPICA DI ACTH
La sindrome di Cushing da secrezione ectopica di ACTH è caratterizzata dalla produzione di ACTH da parte di tumori extraipofisari condizionante una secrezione eccessiva di cortisolo da parte delle ghiandole surrenali.
La dizione “ectopica” definisce la produzione di ACTH da parte di tessuti non propriamente deputati alla secrezione di ACTH, a differenza dell’ipofisi anteriore, organo produttore di ACTH per eccellenza, da cui derivano tumori “eutopici” secernenti ACTH, i.e. la malattia di Cushing. Data la varietà di tumori che possono secernere ACTH, questa sindrome presenta una fisiopatologia complessa ed una presentazione clinica alquanto variabile. L’avanzamento delle conoscenze degli ultimi anni ha consentito di delineare con maggiore precisione diversi aspetti cellulari e molecolari di questa patologia, ad esempio la secrezione di precursori dell’ACTH oppure i fattori coinvolti nell’espressione ectopica del gene per la proopiomelanocortina (POMC), come pure di meglio la classificazione anatomopatologica di questi tumori. Per contro, non vi sono stati progressi significativi sul versante diagnostico e terapeutico, che risulta infatti laborioso e talora insoddisfacente. Questa sindrome continua quindi a presentare un serio problema clinico ma, al tempo stesso, a fornire importanti spunti fisiopatologici. (Prenota una visita endocrinologica).
ANATOMIA PATOLOGICA
La prima classificazione dei tumori neuroendocrini risale al 1968 quando Pearse riconobbe la capacità di cellule neuroendocrine di captare e decarbossilare i precursori delle amine biogene, da qui il concetto di APUD (Amine Precursor Uptake and Decarboxylation). La capacità di queste cellule di elaborare le amine biogene si traduce a livello istologico nella presenza dei tipici granuli secretori situati presso la membrana plasmatica, una caratteristica a tutt’oggi ritenuta indispensabile per stabilire la diagnosi di tumore neuroendocrino. In tempi successivi la classificazione di questi tumori si è basata sull’origine embrionale e sui peptidi prodotti dal tessuto neoplastico mentre secondo la classificazione più recente i tumori vengono suddivisi in base al grado di differenziazione istologica, all’angioinvasività, ai parametri di proliferazione cellulare e alle dimensioni della neoplasia [1]. Si riconoscono quindi tumori neuroendocrini ben differenziati, tra cui figurano i carcinoidi “tipici” ad andamento benigno ed i carcinoidi “atipici” a basso grado di malignità, i carcinomi neuroendocrini scarsamente differenziati ad alto grado di malignità che possono essere a piccole cellule (i.e. microcitomi) oppure a cellule grandi o intermedie ed i tumori misti esocrini-endocrini. Questa classificazione riveste anche un’importanza clinica e diagnostica, oltre che terapeutica, in quanto i carcinoidi benigni presentano un andamento lento e paucisintomatoco, e, in caso di produzione di ACTH, una secrezione ormonale che rassomiglia a quella ipofisaria; per contro, i carcinomi neuroendocrini risultano più aggressivi e la secrezione di ACTH più svincolata dai normali meccanismi regolatori (vedi avanti). I tumori neuroendocrini più frequentemente responsabili della sindrome da secrezione ectopica di ACTH sono i carcinoidi bronchiali o timici, il microcitoma polmonare, i tumori delle isole pancreatiche, il carcinoma midollare della tiroide ed il feocromocitoma. (Prenota un’ecografia tiroidea). Più raramente, la sindrome può essere attribuita a carcinoidi dell’apparato gastroenteropancreatico oppure a isole di tessuto tumorale come “pulmonary tumorlets”.
Oltre ai tumori neuroendocrini propriamente detti, la sindrome di secrezione ectopica di ACTH può essere dovuta alla differenziazione neuroendocrina di un tumore maligno di organi non propriamente neuroendocrini, come il rene o l’osso. In questi casi, si presume che la presenza di cellule neoplastiche neuroendocrine rappresenti l’amplificazione di proprietà istologiche intrinseche della popolazione cellulare come conseguenza della degenerazione neoplastica. E’ noto, infatti, come quasi tutti gli organi non endocrini alberghino cellule neuroendocrine che producono, in condizioni fisiologiche, solo piccole quantità di precursori peptidici per lo più inattivi. La loro trasformazione neoplastica porterebbe per contro alla produzione ed al rilascio di maggiori quantità di ormoni, con la possibilità di indurre una sindrome paraneoplastica. Sono stati descritti pazienti affetti da sindrome da secrezione ectopica di ACTH dovuta a neoplasie prostatiche, epatiche, mammarie, ovariche, appendicolari, della corteccia surrenalica, del pancreas esocrino ed addirittura dei seni paranasali. (Prenota una visita endocrinologica).
BIOLOGIA MOLECOLARE
L’ACTH, come è noto, deriva da un precursore peptidico di 241 aminoacidi, la proopiomelanocortina, a sua volta derivata da un polipeptide di 267 aminoacidi, la prePOMC. La POMC di per sé non possiede attività biologica mentre i suoi derivati: ACTH, endorfine e ormoni melanocito-stimolanti (MSH), svolgono funzioni essenziali per la sopravvivenza.
Il gene per la POMC viene espresso in maniera variabile nei vari tessuti a seconda del promotore da cui inizia la trascrizione genica. Si possono così riscontrare due principali specie di mRNA, una di 1,1 kb, comprensiva di tutti e tre gli esoni del gene per la POMC, e una di 0,8 kb che corrisponde al solo esone 3 del gene per la POMC. Il frammento da 0,8 kb viene espresso quasi ubiquitariamente nell’organismo, mentre il frammento da 1,1 kb si riscontra solo nei tessuti che secernono di derivati peptidici della POMC. Questa discrepanza dipende dal fatto che il frammento di mRNA breve codifica solo per la parte carbossiterminale del peptide mentre non comprende il peptide segnale della prePOMC, essenziale per la trasduzione della catena peptidica nascente nel reticolo endoplasmatico rugoso (REG). La POMC, infatti, viene elaborata all’interno del REG da enzimi specifici, le prohormone convertasi PC1 e PC2, per produrre ad opera della PC1 l’ACTH, il peptide N-terminale, il joining peptide e la ß lipotropina e, dopo un’ulteriore elaborazione enzimatica ad opera della PC2, l’a, il ß ed il gMSH, il CLIP, la g lipotropina e l’a, la ß e la g endorfina. A seconda del corredo enzimatico di una cellula neuroendocrina essa sarà quindi capace di elaborare la POMC per ottenere ACTH o altri peptidi.
Due considerazioni rivestono particolare importanza nei tumori neuroendocrini secernenti ACTH. Una prima di ordine molecolare riguarda il fatto che l’espressione del gene per la POMC al di fuori dell’ipofisi è, come già detto, limitata al frammento di mRNA breve. Pertanto, per avere la secrezione dei peptidi derivati dalla POMC si suppone avvenga un processo di “promoter switching” con l’attivazione del promotore a monte dell’esone 1 (solitamente inattivo in questi tessuti) e la trascrizione del frammento completo di mRNA da 1,1 kb. In alternativa, in alcuni carcinomi neuroendocrini è stata descritta una variante di mRNA da 1,3 kb originata da un promotore situato in 5’. Questo mRNA dà luogo da un polipeptide che contiene il peptide segnale e può quindi essere trasdotto nel REG. Un’altra considerazione ruiguarda la capacità di elaborare il peptide POMC. Infatti, non tutti i tessuti neuroendocrini possiedono le prohormone convertasi necessarie per digerire il precursore. In base al suo corredo enzimatico quindi, il tumore potrà secernere la POMC stessa, l’ACTH oppure altri frammenti POMC-derivati. Questi ultimi non possiedono attività steroidogenica e pertanto è necessaria la liberazione di ACTH da parte del tumore per indurre il quadro di ipercortisolismo. La secrezione dei derivati della POMC riveste peraltro un significato diagnostico e prognostico; livelli elevati di POMC sono stati riscontrati nei carcinomi neuroendocrini scarsamente differenziati mentre risultano per lo più nella norma nei tumori neuroendocrini ben differenziati [2].
Anche la regolazione del gene per la POMC varia a seconda del tessuto dove il gene viene espresso. Nell’ipofisi anteriore, come ben noto, la sintesi di POMC viene stimolata dal corticotropin-releasing hormone (CRH) e dalla vasopressina (AVP) ed inibita dai glicocorticoidi. Questa regolazione appare per lo più conservata nei tumori neuroendocrini ben differenziati mentre viene a mancare nei carcinomi neuroendocrini. L’insensibilità nel tessuto tumorale ai glicocorticoidi può essere attribuita a mutazioni del gene del recettore per i glicocorticoidi che rendono inattive oppure ne impediscono la trascrizione e sintesi. Per contro, è stato dimostrato che la conservata responsività al CRH è legata alla sintesi intratumorale del CRH stesso e quella alla desmopressina (DDAVP), un analogo dell’AVP, dipende dall’espressione tissutale del recettore V3 dell’AVP. [3]
CLINICA
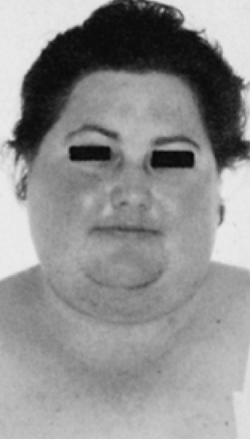
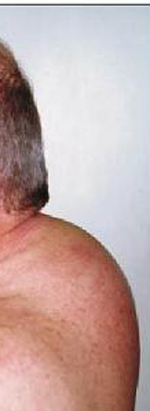
La sindrome da secrezione ectopica di ACTH dà luogo ad un quadro in genere conclamato di ipercortisolismo: obesità tronculare, “facies lunaris”, “gibbo di bufalo”, irsutismo, acne, strie rubre ai fianchi, ipertensione arteriosa, diabete mellito, miopatia degli arti superiori ed inferiori, oligo-amenorrea, disturbi della sfera psichica, perdita della libido, osteoporosi e infezioni ricorrenti. Il quadro clinico presenta solitamente un’insorgenza graduale, se responsabile della sindrome è un tumore neuroendocrino ben differenziato mentre un esordio rapido con manifestazioni cliniche imponenti caratterizza per lo più i carcinomi neuroendocrini scarsamente differenziati. Questi ultimi pazienti, inoltre, possono presentare una spiccata ipokaliemia associata ad alcalosi metabolica, così come un’iperpigmentazione dovuta agli alti livelli di ACTH e di peptidi derivati dalla POMC. Alle manifestazioni dell’ipercortisolismo si possono ovviamente affiancare quelle legate al tumore neuroendocrino di per sè, dovute alla massa neoplastica o alla secrezione di altri peptidi o amine. (Prenota una visita endocrinologica).
{kind=link}
{kind=link}
{kind=link}
DIAGNOSI
La diagnosi di questa sindrome di basa anzitutto sul riconoscimento della presenza di ipercortisolismo. A questo scopo, sono determinanti il dosaggio del cortisolo libero urinario delle 24 ore, la determinazione del cortisolo plasmatico alla mezzanotte e l’inibizione dei valori mattutini di cortisolo dopo la somministrazione di 1 mg desametasone la mezzanotte precedente. L’esito di queste prove è solitamente univoco nei pazienti con sindrome da secrezione ectopica di ACTH, con il riscontro di valori per lo più molto elevati di cortisolo urinario e plasmatico, la mancanza del ritmo nictemerale del cortisolo plasmatico e della sua sopprimibilità da parte del desametasone [4]. Il dosaggio dell’ACTH, eseguito in seconda battuta, consente di confermare la natura ACTH-dipendente della patologia, escludendo pertanto una sindrome di Cushing di origine primitivamente surrenalica. I pazienti con sindrome da secrezione ectopica di ACTH presentano solitamente concentrazioni plasmatiche di ACTH ai limiti superiori della norma, oppure, come spesso accade nei carcinomi neuroendocrini, francamente elevate. In alcuni centri è possibile misurare direttamente le concentrazioni plasmatiche di POMC le quali, come già menzionato, possono risultare elevate nella sindrome da ACTH ectopico, anche se questo riscontro non risulta indispensabile per formulare la diagnosi. Il passaggio diagnostico più difficoltoso resta invece la diagnosi differenziale con la malattia di Cushing, ossia con un adenoma ipofisario secernente ACTH. Questo passaggio si avvale di tre principali prove diagnostiche, nessuna della quali offre un’accuratezza diagnostica assoluta. La prova diagnostica attualmente utilizzata per prima è il test di stimolo con CRH, prova altamente sensibile e specifica che nella sindrome da ACTH ectopico non riesce in genere ad evocare un’elevazione dei livelli plasmatici di ACTH e cortisolo. Meno attendibile risulta l’altro test dinamico, l’inibizione del cortisolo con desametasone ad alte dosi, in quanto una percentuale considerevole di pazienti con questa sindrome presenta un’inibizione del cortisolo con questo test [4]. Se ambedue le prove citate indicano un’origine ipofisaria della patologia, non sono indispensabili altre indagini ormonali ed il paziente può essere inviato al neurochirurgo dopo l’esecuzione della risonanza magnetica nucleare della regione sellare. Merita qui ricordare come quest’ultima risulta negativa nel 40% circa dei pazienti portatori di un microadenoma ipofisario secernente ACTH. Se invece i risultati del test al CRH e del test con desametasone ad alte dosi danno risultati contrastanti o ambedue non indicativi di un’origine ipofisaria, diventa necessario eseguire il cateterismo dei seni petrosi inferiori con dosaggio dell’ACTH prima e dopo stimolo con CRH al fine di escludere la presenza di un tumore ipofisario. Il riscontro di un gradiente centro-periferia delle concentrazioni plasmatiche di ACTH uguale o superiore a 2 in condizioni basali e uguale o superiore a 3 dopo stimolo con CRH, esclude con certezza una sindrome da secrezione ectopica di ACTH indirizzando quindi la diagnosi verso la malattia di Cushing [5]. A rendere più complesso l’iter diagnostico per la patologia in questione vi sono ragioni di ordine epidemiologico da tenere in considerazione. Poiché la sindrome da secrezione ectopica di ACTH è nettamente meno frequente della malattia di Cushing e nessuna delle indagini diagnostiche oggi disponibili ha un’accuratezza diagnostica assoluta, un risultato indicativo di una sindrome da secrezione ectopica ha più probabilità di rappresentare un falso negativo nell’ambito di una malattia di Cushing che di riflettere realmente la prima diagnosi. Tutto questo va tenuto in grande considerazione nelle decisioni diagnostiche delle forme di ipercortisolismo ACTH-dipendente. (Prenota una visita endocrinologica).
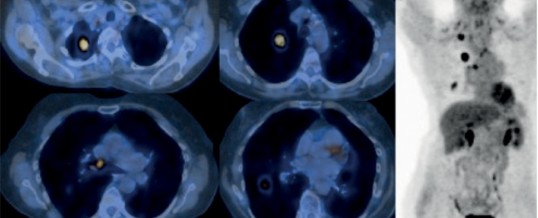
La fase finale dell’iter diagnostico della sindrome da secrezione ectopica di ACTH è rappresentata dall’identificazione e localizzazione del tumore, avvalendosi della TAC, della risonanza o di metodiche scintigrafiche. Queste indagini sono superflue se l’esistenza e la sede del tumore sono già note mentre sono necessarie e possono risultare complesse nel caso di tumori neuroendocrini di piccole dimensioni. Talora la neoplasia può non essere visualizzata con alcuna delle moderne tecniche radiologiche, inclusa la scintigrafia con analoghi marcati della somatostatina (octreoscan); in questi casi la sindrome viene definita da tumore neuroendocrino “occulto”.
TERAPIA
L’orientamento terapeutico viene determinato dalla stadiazione della neoplasia e dalla gravità dell’ipercortisolismo. Il primo approccio al tumore neuroendocrino è solitamente chirurgico, specie nelle forme ben differenziate. Se l’exeresi della neoplasia ha avuto successo, i livelli di ACTH si abbasseranno subito dopo l’intervento causando un quadro di iposurrenalismo che va compensato con un’adeguata terapia steroidea sostitutiva. Il monitoraggio dei livelli di ACTH può servire nel follow-up del paziente per riconoscere lo sviluppo di metastasi o eventuali recidive. Se la massa tumorale è estesa oppure la presenza di metastasi diffuse preclude l’intervento chirurgico, la strategia terapeutica dovrà ripiegare sul trattamento antiblastico eventualmente associato ad interferone e/o analoghi della somatostatina a lunga durata d’azione, sulla chemoembolizzazione delle arterie epatiche o, come proposto con successo in tempi recenti, sulla terapia radiometabolica con analoghi marcati della somatostatina.
Se il tumore neuroendocrino è occulto oppure l’ipercortisolismo compromette gravemente le condizioni cliniche del paziente, si deve procedere alla correzione dell’ipercortisolismo prima di rimuovere il tumore. L’evoluzione delle tecniche chirurgiche laparoscopiche consente oggi di effettuare con successo una surrenectomia bilaterale anche in pazienti molto compromessi [6]. In alternativa, si può procedere ad una surrenectomia farmacologica utilizzando farmaci ad azione rapida come l’etomidato oppure ricorrere all’antagonista del recettore per i glicocorticoidi, il mifepristone. La surrenectomia bilaterale trova anche indicazione in presenza di un tumore neuroendocrino occulto o comunque non asportable, anche se il quadro di ipercortisolismo non presenta particolare gravità, non essendo pensabile di impostare un trattamento permanente con inibitori della steroidogenesi. Sono stati peraltro descritti pazienti in cui il tumore occulto si è manifesto a 10 anni dalla surrenectomia, consentendo la sua rimozione. (Prenota una visita chirurgica).
In conclusione, la sindrome da secrezione ectopica di ACTH è una patologia complessa che riconosce diverse eziologie, un iter diagnostico spesso laborioso ed un approccio terapeutico non sempre soddisfacente. Grande pertanto è l’interesse e la necessità di un avanzamento delle conoscenze per questa patologia, al fine migliorare la prognosi di questi pazienti.
Dott.ssa Francesca Pecori Giraldi
Bibliografia
1) Solcia E, Kloppel G, Sobin LH, Capella C, Heitz PU. Histological typing of endocrine tumours. International Histological Classification of Tumors. World Health Organization, Springer Verlag, Berlin, 2000.
2) Raffin-Sanson ML, Massias JF, Dumont C, Raux-Demay MC, Proeschel MF, Luton JP, Bertagna X. High plasma proopiomelanocortin in aggressive adrenocorticotropin-secreting tumors. Journal of Clinical Endocrinology & Metabolism 1996; 81:4272-7.
3) de Keyzer Y, Lenne F, Auzan C, Jégou S, René P, Vaudry H, Kuhn JM, Luton JP, Clauser E, Bertagna X. The pituitary V3 vasopressin receptor and the corticotroph phenotype in ectopic ACTH syndrome. Journal of Clinical Investigation 1996; 97: 1311-8.
4) Invitti C, Pecori Giraldi F, De Martin M, Cavagnini F, the Study Group of the Italian Society of Endocrinology on the Pathophysiology of the Hypothalamic-Pituitary-Adrenal Axis. Diagnosis and management of Cushing’s syndrome: results of an Italian multicentre study. Journal of Clinical Endocrinology & Metabolism 1999; 84: 440-8.
5) Oldfield EH, Doppman JL, Nieman LK, Chrousos GP, Miller DL, Katz DA, Cutler Jr. GB, Loriaux DL. Petrosal sinus sampling with and without corticotropin-releasing hormone for the differential diagnosis of Cushing’s syndrome. New England Journal of Medicine 1991; 325: 897-905.
6) Vella A, Thomson GB, Grant CS, van Heerden JA, Farley DR, Young WF Jr. Laparoscopic adrenalectomy for adrenocorticotropin-dependent Cushing’s syndrome. Journal of Clinical Endocrinology & Metabolism 2001; 86: 1596-9.
7) Cavagnini F, Putignano P, Pecori Giraldi F. Ectopic ACTH syndrome. In: Giustina A. (ed). Neuroendocrine Tumors on CD-ROM, Beaufour-Ipsen, Chap. 4, 2002.
AGO
2011