SINDROME DI CUSHING
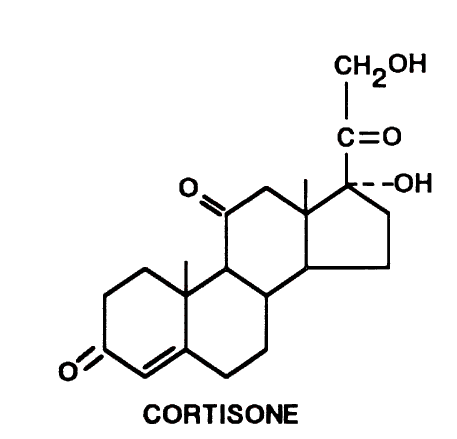
Sindrome di Cushing (ipercortisolismo) è il termine con cui si definisce il quadro clinico che fa seguito ad una prolungata esposizione ad elevati livelli di cortisonici nel sangue. Il cortisone in eccesso presente nell’ipercotisolismo può essere prodotto dal nostro organismo (cortisolo) oppure essere assunto dall’esterno (prednisone, desametasone etc.).
L’assunzione di farmaci contenenti cortisone è la causa più frequente di sindrome di Cushing in quanto la maggior parte dei pazienti sottoposti ad una terapia cronica con cortisonici, infatti, sviluppano una sindrome di Cushing da farmaci (iatrogena). Per contro, la sindrome di Cushing endogena, dovuta ad un eccessiva produzione di cortisolo da parte di due ghiandole poste al di sopra dei reni (surreni), è un’evenienza decisamente più rara.
{kind=link}
{kind=link}
{kind=link}
CLINICA
La sindrome di Cushing si presenta con un quadro clinico caratterizzato da:
Obesità
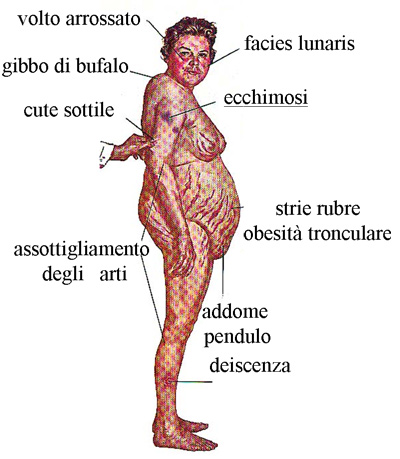
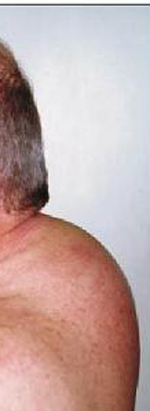
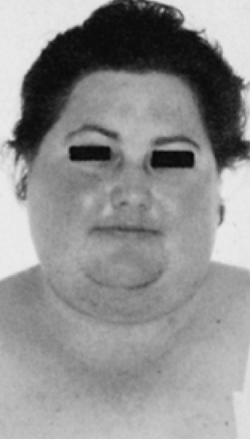
L’obesità è solitamente di grado lieve-moderato e raggiunge raramente il III grado come invece può avvenire nell’obesità classica; questo perché nella sindrome di Cushing l’obesità è per lo più di tipo tronculare (cioè il grasso in eccesso si concentra prevalentemente sul tronco e non sugli arti che paiono spesso più sottili). Inoltre il tessuto adiposo si deposita in sedi caratteristiche, come la regione cervicale (gibbo di bufalo), le fossette sovraclaveari e attorno alle guance e nella regione temporale, determinando in tal modo la classica faccia lunare. Il grasso, inoltre, può depositarsi anche a livello orbitario, favorendo la comparsa di esoftalmo, e nello spazio epidurale contribuendo allo sviluppo di deficit neurologici. (Prenota una visita neurologica).
Miopatia e atrofia muscolare
A fronte della deposizione di grasso al tronco ed al volto, colpisce il marcato assottigliamento degli arti superiori ed inferiori e del cingolo scapolare. La miopatia e l’ipotrofia dei tessuti muscolari sono, infatti, fra gli aspetti più caratteristici della sindrome di Cushing, e sono dovuti all’atrofia delle fibre muscolari, ed alla ridotta produzione di proteine indotta dall’eccesso di glicocorticoidi. Questa sofferenza muscolare si traduce nella difficoltà ad effettuare alcuni movimenti specifici, quali alzarsi dalla sedia senza appoggio.
Osteoporosi
L’osteoporosi è quella condizione caratterizzata da una ridotto contenuto di calcio nelle ossa, attualmente diagnosticabile anche con metodiche senza radiazioni come la Densitometria REMS, che pertanto risultano più fragili e più soggette a fratture. Nella sindrome di Cushing l’osteoporosi si osserva molto più frequentemente che nella norma.
Alterazioni cutanee
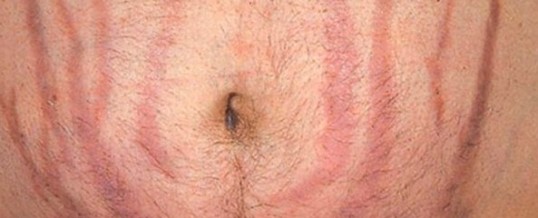
La sindrome di cushing si accompagna spesso ad assottigliamento della cute che lascia così trasparire le strutture vascolari sottostanti, presentandosi “rubizza”, specie nelle zone esposte alla luce. Il volto appare quindi “pletorico” oltre che arrotondato per l’accumulo di adipe, assumendo il tipico aspetto a luna piena. L’alterazione cutanea più caratteristica, anche se non obbligatoria, della sindrome di Cushing è rappresentata dalle strie rubrae, smagliature profonde, di colore rosso-violaceo, localizzate sull’addome, ai fianchi, alla radice degli arti e sui quadranti laterali delle mammelle. Queste smagliature vanno distinte da quelle che insorgono dopo il parto o in seguito ad un dimagramento rapido, che sono solitamente più superficiali e meno pigmentate. Nei pazienti con sindrome di Cushing sono frequenti sudorazione maleodorante (seborrea), acne al volto e al tronco, ed eccessiva crescita di peli (irsutismo). Infine, nelle forme di sindrome di Cushing da secrezione ectopica di ACTH la cute può apparire più scura ed iperpigmentata per l’attivazione dei melanociti. (Prenota una visita dermatologica).
Alterazioni psichiatriche
Nel paziente con sindrome di Cushing l’eccesso di cortisolo e/o ACTH determina instabilità e depressione del tono dell’umore, ansietà, facilità agli scatti d’ira e agli attacchi di panico, insonnia e, più raramente, psicosi maniacali o pensieri suicidari. (Prenota una valutazione psicologica).
Alterazioni sessuali
Il GnRH è un ormone che viene secreto in modo pulsatile dall’ipotalamo e che con la sua pulsatilità regola la funzione sessuale. I glicocorticoidi interferiscono con la secrezione pulsatile di GnRH e quindi determinano riduzione della vis nell’uomo, irregolarità mestruali nella donna (amenorrea) e riduzione della libido e della fertilità in entrambi i sessi.
Alterazioni cardiovascolari
L’ipertensione arteriosa si riscontra nel 75% dei pazienti con sindrome di Cushing. L’aumento dei valori pressori può avere conseguenze avverse sulla funzione cardiaca, così come sul sistema vascolare, favorendo l’ispessimento e la rigidità delle pareti dei vasi del sangue. Queste alterazioni, insieme all’aumentata incidenza di diabete mellito, costituiscono fattori di rischio importanti per eventi cardio- e cerebrovascolari (infarti, ictus) che rappresentano, infatti, la principale causa di morte dei pazienti con sindrome di Cushing. (Prenota una visita cardiologica).
Infezioni
I pazienti con sindrome di Cushing sono maggiormente predisposti a sviluppare infezioni da germi opportunisti (funghi) oppure alla riattivazione di infezioni pregresse, come la tubercolosi.
Altre alterazioni
Tra le altre complicanze a cui è esposto il paziente con sindrome di Cushing, sono da menzionare quelle oculari (glaucoma) e quelle renali (calcoli renali). Infine, l’eccesso di cortisolo provoca una serie di alterazioni metaboliche, le più frequenti rappresentate dal diabete mellito e dalla presenza di valori elevati di colesterolo e trigliceridi (dislipidemia). Nel paziente con sindrome di Cushing si osserva anche una aumentata frequenza di valori bassi di potassio (ipokaliemia) ed elevati di calcio (ipercalcemia).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
CLASSIFICAZIONE
Esclusa la forma più frequente (ovvero quella conseguente l’assunzione di farmaci), nell’ambito della la sindrome di Cushing si distinguono due forme: forme ACTH-dipendenti ed ACTH-indipendenti a seconda della a causa che determina l’esagerata produzione di cortisone:
CUSHING ACTH – DIPENDENTE:
– adenoma ipofisario secernente ACTH;
– secrezione ectopica di ACTH;
– secrezione ectopica di CRH.
CUSHING ACTH – INDIPENDENTE:
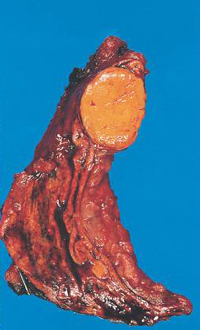
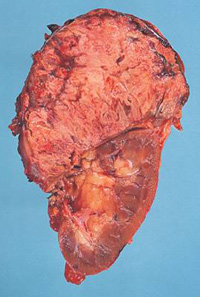
– adenoma e carcinoma surrenalico;
– iperplasia micro e macronodulare dei surreni;
– displasia nodulare pigmentata.
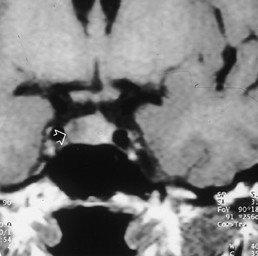
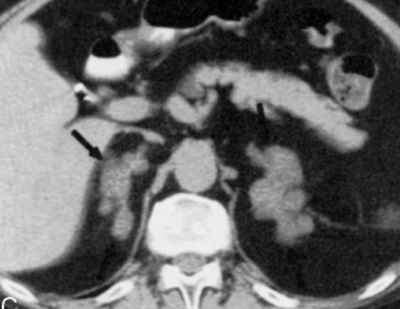
La causa più frequente (70% dei casi) di sindrome di Cushing endogeno è adenoma ipofisario secernente ACTH, ovvero un tumore dell’ipofisi che stimola i surreni a produrre quantità di cortisolo in eccesso rispetto quelle necessarie. Nella maggior parte dei casi il tumore è di piccole dimensioni (microadenoma), spesso così piccolo da non essere visualizzato dalla risonanza magnetica, e solo in un paziente su 10 esso supera il centimetro di diametro (macroadenoma). Circa il 15% delle sindromi di Cushing, invece, deriva dalla secrezione ectopica di ACTH; ovvero alcuni tumori, che normalmente non producono sostanze in grado di stimolare la produzione di cortisolo, incominciano a produrle determinando la sindrome di Cushing. Tra le forme ACTH-indipendenti, infine, ricordiamo i tumori del surrene (adenomi e carcinomi), l’iperplasia surrenalica nodulare e la displasia surrenalica nodulare pigmentata (una condizione rara che si riscontra frequentemente nell’ambito di una sindrome genetica trasmessa nota come complesso di Carney). (Prenota una visita endocrinologica).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
DIAGNOSI
La diagnosi è spesso difficile e complessa. Per prima cosa bisogna dimostrare che ci si trova davanti ad una vera sindrome di Cushing e non ad una di quelle condizioni note come pseudocushing (obesità, alcoolismo e depressione) che possono essere facilmente confuse con essa. Per far ciò vengono eseguiti degli esami specifici come la ricerca del cortisolo libero urinario nelle urine delle 24 ore, il dosaggio del cortisolo plasmatico a mezzanotte o al mattino dopo inibizione con basse dosi di desametasone (test di Nugent). Una volta appurato che si tratta veramente di una sindrome di Cushing bisogna distinguere se si tratta di una forma ACTH dipendente o indipendente e per questo è opportuno dosare i valori di ACTH. Nei casi più complicati può essere necessario eseguire accertamenti radiologici (risonanza magnetica nucleare) e soprattutto test specifici di stimolazione o di inibizione come: il test al CRH, il test di inibizione con alte dosi di desametasone (test di Liddle) ed il cateterismo dei seni petrosi. (Prenota una visita endocrinologica).
TERAPIA
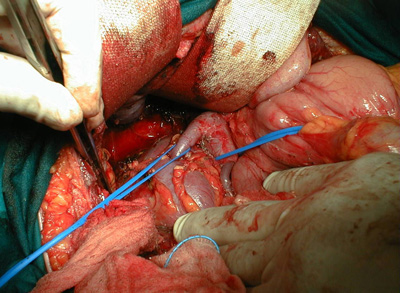
La terapia ideale e definitiva consiste nell’asportare il tumore che determina l’aumentata produzione di cortisolo. Nel caso dell’adenoma ipofisario l’intervento viene effettuato dal neurochirurgo per via trans-naso-sfenoidale, negli altri casi, invece, interviene l’endocrino-chirurgo od il chirurgo generale asportando il tumore che causa l’ipercortisolismo quale che sia la sua localizzazione. Tuttavia, nei casi in cui vi è una recidiva della malattia dopo l’intervento ed in tutti quei casi (non rarissimi) in cui non è stato possibile individuare la causa del tumore può essere necessario l’uso di farmaci surrenostatici (che riducono la produzione di cortisolo da parte dei surreni). Altri farmaci, i.e. doplaminergici, acido retinoico e pasireotide (SOM230), sono attualmente in studio come opzioni terapeutiche. Tutti i suddetti farmaci, tuttavia, a volte, possono risultare comunque inefficaci nel controllare l’ipercortisolismo e quindi, pur di controllare l’ipercortisolismo, può essere necessario intervenire chirurgicamente asportando i surreni (surrenectomia) che sono gli unici organi del nostro corpo in grado di produrre cortisolo. In rari casi, tuttavia, si può sviluppare la sindrome di Nelson.
{kind=link}
Prenota una visita specialistica endocrinologica in merito a questo argomento.
Prof. Francesco Cavagnini
e
Dott. Massimiliano Andrioli
Specialista in Endocrinologia e Malattie del Ricambio
Centro EndocrinologiaOggi, Roma
viale Somalia 33A, Roma
tel/fax 0686391386
cell 3337831426
Studio EndocrinologiaOggi, Lecce
via Ruffano 4, Casarano (Lecce)
tel/fax 0686391386
cell 3337831426
Bibliografia
1) Ross EJ, Linch Dc. Cushing’s syndrome-killing disease: discriminatory value of signs and symptoms aiding early diagnosis. Lancet 1982; 2: 646-649.
2) Jones K.L. The Cushing syndromes. Pediatric Clinics of North America. Vol.37, No 6, December 1990
3) Colao A, Pivonello R, Spiezia S et al. Persistence of increased cardiovascular risk in patients with Cushing’s disease after five years of successful cure. J Clin Endocrinol Metab; 84:2664-2672.
4) Cavagnini F, Pecori Giraldi F. Adrenal causes oh hypercortisolism. Endocrinology 5th Ed. DeGroot Lj, & Jameson JL, Elsevier Philadelphia, Chap. 122 (in press) 2005.
GIU
2011