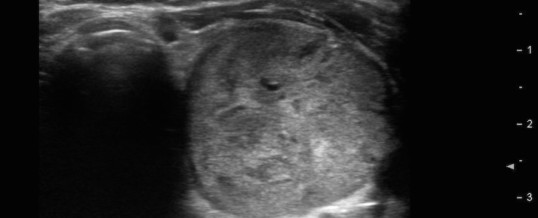
CARCINOMA MIDOLLARE TIROIDE
Il carcinoma midollare della tiroide (CMT) origina dalle cellule parafollicolari o cellule C della tiroide e rappresenta circa il 5-10% di tutti i carcinomi tiroidei.
Le cellule C migrano durante la vita embrionale dalla cresta neurale alla ghiandola tiroidea, sono localizzate nello strato basale dei follicoli tiroidei e rappresentano l’1% delle cellule tiroidee.
Le cellule C sono distribuite in tutta la ghiandola tiroidea ma sono più concentrate nella zona tra il terzo superiore e i due terzi inferiori dei lobi. Si distinguono dalle cellule dell’epitelio follicolare soprattutto per la capacità di secernere la calcitonina (CT), un piccolo peptide a singola catena costituito di 32 aminoacidi il cui gene è localizzato sul braccio corto del cromosoma 11. Nel sangue sono presenti varie forme di CT e la concentrazione normale di CT sierica è inferiore a 10 pg/ml. Livelli basali elevati di CT, oltre che nel CMT, si possono riscontrare in alcune condizioni fisiologiche (gravidanza, dopo esercizio fisico intenso, alcoolismo) e patologiche (insufficienza renale cronica, tiroidite cronica autoimmune, nel 15% dei pazienti con tumori neuroendocrini).
Il test di stimolo con pentagastrina è quello più utilizzato nella pratica clinica per stimolare la secrezione di CT e discriminare tra ipercalcitoninemie neoplastiche e non. Occasionalmente pazienti con iperplasia delle cellule C o con microcarcinoma midollare possono presentare valori di CT basali solo lievemente elevati ma che possono aumentare significativamente dopo stimolazione con pentagastrina.
Il CMT nella maggior parte dei casi si manifesta sotto forma sporadica (80%) o ereditaria (20%). I pazienti con carcinoma midollare sporadico della tiroide si presentano generalmente con un nodulo tiroideo associato, in circa la metà dei casi, alla presenza di metastasi linfonodali nella regione del collo. Raramente sono presenti al momento della diagnosi metastasi a distanza e, in questi casi, gli organi generalmente più colpiti sono il fegato, i polmoni e le ossa.
La presenza di una sintomatologia caratterizzata da alvo diarroico o da flush cutanei al volto associati alla presenza di un nodulo tiroideo, sono caratteristiche cliniche che devono indurre il medico a sospettare la presenza di un carcinoma midollare.
La totalità dei pazienti con carcinoma midollare clinicamente manifesto presentano concentrazioni basali elevate di CT. Il dosaggio della CT, ormone che è sempre consigliabile eseguire in presenza di noduli tiroidei, nel caso del carcinoma midollare ha una attendibilità diagnostica maggiore a quella dell’esame citologico su agoaspirato tiroideo (prenota un agoaspirato tiroideo), il quale non sempre risulta dirimente in tutti i casi e può richiedere la conferma della diagnosi attraverso l’esame immunoistochimico per CT.
Circa il 4-5% di casi di carcinoma midollare sporadico sono in realtà di tipo ereditario. Risulta quindi necessario anche nei casi apparentemente sporadici, eseguire lo screening genetico per individuare forme ereditarie erroneamente diagnosticate come sporadiche.
La forma ereditaria del carcinoma midollare si trasmette con carattere autosomico dominante a causa di una mutazione germinale del proto-oncogene RET codificante per un recettore tirosino chinasico (prenota anlaisi genetica RET). Può manifestarsi clinicamente solo con la presenza di un carcinoma midollare o nell’ambito di una sindrome endocrina multipla coinvolgente altre ghiandole (MEN 2).
Lo screening genetico permette di identificare i familiari di un soggetto affetto, portatori anch’essi del gene mutato ma inconsapevoli della loro malattia e destinati a sviluppare il carcinoma midollare della tiroide. In questi soggetti è quindi possibile intervenire precocemente con la terapia chirurgica tiroidea profilattica o precoce che consente, rispettivamente, la prevenzione della malattia o la guarigione se già manifesta in forma subclinica. Lo screening deve essere eseguito in tutti i familiari di primo grado di un soggetto affetto.
La MEN II A è una sindrome in cui si associano il carcinoma midollare, il feocromocitoma (10-60%) e l’iperparatiroidismo (10-25%). Il carcinoma midollare in questi pazienti raramente si manifesta prima dei 10 anni e la sua prevalenza aumenta con l’aumentare dell’età. In alcune famiglie affette da MEN IIA è stata descritta una lesione della cute iperpigmentata ed intensamente pruriginosa, detta lichen cutaneo amiloidosico, localizzata nella regione interscapolare; può verificarsi molto precocemente e spesso precede la manifestazione clinica del carcinoma midollare. (Prenota una visita endocrinologica).
La MEN II B è una sindrome caratterizzata dall’associazione di carcinoma midollare, feocromocitoma, ganglioneuromatosi (che include i neurinomi mucosi evidenti nella parte distale della lingua, nella mucosa delle labbra e dell’intero tratto gastro-intestinale ed eventualmente urinario), habitus marfanoide (arti lunghi e sottili, lassità dei legamenti e inversione del rapporto tra la parte superiore ed inferiore del corpo) ed anomalie scheletriche (alterazioni delle epifisi femorali, petto scavato e facies caratteristica). Di fronte ad un paziente con aspetto marfanoide ed una facies tipica con classici neurinomi, associati o meno a disordini gastro-intestinali, il medico dovrebbe immediatamente sospettare la diagnosi di MEN IIB. Il carcinoma midollare che si manifesta in questa sindrome è una forma molto aggressiva, che si manifesta precocemente (generalmente prima dei 10 anni di età) ed è associato frequentemente a metastasi linfonodali e a distanza. (Prenota una visita endocrinologica).
{kind=link}
STAGING PRE-OPERATORIO
I pazienti con carcinoma midollare sia sporadico che ereditario prima dell’intervento chirurgico di tiroidectomia devono essere valutati per la presenza di un eventuale feocromocitoma mediante ecografia addominale e dosaggio di metanefrine o catecolamine urinarie. Anche la presenza di iperparatiroidismo primitivo deve essere esclusa mediante il dosaggio della calcemia e del paratormone. Lo staging pre-operatorio comprende inoltre l’ecografia della tiroide e del collo, con particolare attenzione alle catene linfonodali. Eventuali linfonodi sospetti devono essere sottoposte ad agoaspirato linfonodale con dosaggio della calcitonina. Indicata l’esecuzione di una Tac collo-torace-addome e della scintigrafia ossea per escludere la presenza di metastasi.
TRATTAMENTO
Il trattamento terapeutico di scelta nei pazienti affetti da carcinoma midollare sporadico e familiare è di principio la tiroidectomia totale associata a svuotamento linfonodale del compartimento centrale. La dissezione degli altri compartimenti linfonodali dovrebbe essere eseguita solo se coinvolti clinicamente. (Prenota una visita chirurgica). Le metastasi linfonodali si verificano nel 10% dei casi con lesioni primitive di diametro inferiore ad un centimetro e fino al 90% dei casi con tumore evidente clinicamente.
Nelle forme ereditarie che non hanno una malattia clinicamente evidente, la tiroidectomia totale profilattica associata alla dissezione del compartimento centrale può essere risolutiva se eseguita quanto più precocemente. E’ tuttavia ancora controverso se la dissezione del compartimento centrale debba essere eseguita di routine nei soggetti giovani portatori della mutazione ma senza alcuna evidenza clinica né biochimica di malattia (CT basale e dopo stimolo con pentagastrina indosabile).
Nei pazienti con feocromocitoma accertato (elevati valori di metanefrine o catecolamine urinarie) l’intervento di asportazione del feocromocitoma deve precedere quello di tiroidectomia mediante surrenectomia monolaterale, se la ghiandola surrenalica affetta è unica, monitorando nel tempo la funzione e la morfologia della ghiandola controlaterale. I pazienti con feocromocitoma bilaterale devono ovviamente essere trattati con adrenalectomia bilaterale.
Nel caso di evidente iperparatiroidismo primitivo, la paratiroide sede di un adenoma dovrebbe essere rimossa; se invece è presente una iperplasia diffusa di tutte le paratiroidi, tre di esse dovrebbero essere rimosse e una porzione della quarta dovrebbe essere reimpiantata nel muscolo del braccio.
Nel caso di localizzazione metastatica la terapia dipenderà dalla sede delle lesioni.
Il reintervento chirurgico è consigliabile per le metastasi loco-regionali. Per le metastasi a distanza, la terapia chirurgica è indicata nel trattamento di metastasi uniche (epatiche, cerebrali o polmonari) o a livello osseo nei pazienti con grave rischio di complicanze (fratture patologiche o compressione midollare). La radioterapia esterna è indicata per le metastasi ossee che non possono essere trattate chirurgicamente o per le metastasi cerebrali. La loperamide è utilizzata per controllare la sintomatologia diarroica, gli antistaminici per i flush cutanei e gli antidolorifici per la sintomatologia dolorosa.
Il trattamento con lo iodio radioattivo ovviamente non ha alcuna efficacia poiché le cellule C non captano il radioiodio.
Nel caso di metastasi multiple non trattate chirurgicamente, lo schema di polichemioterapia più utilizzato prevede l’utilizzo della doxorubicina e cisplatino anche se i risultati clinici non sono soddisfacenti, 20-30% di risposte paraziali.
Risultati promettenti sembrano invece provenire dalla sperimentazione, attualmente in fase clinica, di farmaci inibenti l’attività tirosino-chinasica che bloccano la trasduzione del segnale e l’effetto mitogenico dell’ oncogene RET.
FOLLOW-UP
Dopo il trattamento chirurgico per carcinoma midollare, deve essere instaurata una terapia sostitutiva con levo-tiroxina, mantenendo i livelli di TSH nell’ambito dei valori normali ed eseguire a distanza di almeno due mesi dalla terapia chirurgica, i dosaggi di CT: se i valori basali di CT risultano indosabili, l’accertamento deve essere completato mediante un test di stimolo con pentagastrina. La sopravvivenza a 10 anni dei pazienti affetti da carcinoma midollare è in genere del 60-70 %. La maggior parte dei pazienti in cui la malattia è identificata precocemente con lo screening genetico presenta remissione clinica. Pazienti con valori di calcitonina indosabili basali e dopo stimolo con pentagastrina in due valutazioni dopo l’intervento chirurgico sono da considerare in remissione completa. Nei pazienti invece che dopo l’intervento chirurgico presentano persistenza di malattia biochimica per la presenza di valori dosabili di CT è necessario identificare la sede della malattia residua. Le tecniche di localizzazione tumorale includono la TC o la RMN per esplorare il collo, il torace e l’addome, l’ecografia del collo e dell’addome (prenota una ecografia) e la scintigrafia ossea. In mancanza di localizzazione, la strategia migliore è l’attesa e la rivalutazione a 6-12 mesi, senza nessun provvedimento terapeutico.
Prof. Furio Pacini
e
Dott. Marco Capezzone
GIU
2011