
IPERPLASIA SURRENALICA CONGENITA (21 IDROSSILASI)
Iperplasia surrenalica congenita è il termine che insieme a sindromi adreno-genitali viene comunemente usato per descrivere un gruppo di patologie autosomiche recessive dovute alla mancanza di uno dei 5 enzimi che intervengono nella sintesi del cortisolo nella corteccia surrenalica (1).
Il difetto enzimatico più frequente è quello che interessa l’enzima 21-idrossilasi e determina oltre il 90-95 % delle iperplasie surrenaliche congenite (1).
Dal primo caso descritto diversi anni fa, sono stati fatti molti passi in avanti nella conoscenza della malattia, sia per quanto riguarda le basi genetiche che per quanto riguarda la corrispondenza genotipo-fenotipo in questa patologia.
La terapia standard attualmente utilizzata permette di ben controllare la patologia ma la scoperta di nuove e più moderne modalità di trattamento, sembrano dare risultati ancora più promettenti (2). Attualmente, inoltre, è possibile effettuare anche una diagnosi precoce, addirittura in epoca prenatale, che consente di instaurare un trattamento altrettanto precoce già durante la gravidanza.
FISIOPATOLOGIA
La sintesi degli ormoni steroidei surrenalici è un processo complesso che a partire dal colesterolo permette la sintesi del cortisolo, dell’aldosterone e degli ormoni sessuali.
Il colesterolo subisce una serie di trasformazioni enzimatiche in tappe successive ed ogni prodotto di sintesi rappresenta il substrato per la sintesi di un altro composto e così via. Si tratta, pertanto, di una vera e propria cascata di reazioni in cui sono coinvolti diversi enzimi (Figura 1).
L’enzima 21-idrossilasi (chiamato anche CYP21 o P450c21) appartiene alla categoria dei citocromi P-450 e, a livello intracellulare, è localizzato nel reticolo endoplasmatico. Esso catalizza la conversione del 17-idrossi progesterone (17OH-P) a 11-desossicortisolo, un precursore del cortisolo, e del progesterone in desossicorticosterone, un precursore dell’aldosterone. (Prenota un test genetico per questo deficit).
Nel caso in cui vi sia un deficit, parziale o totale, nella funzionalità dell’enzima il paziente portatore del difetto non è in grado di sintetizzare efficientemente una adeguata quantità di cortisolo; pertanto, i bassi valori di cortisolo prodotti non sono in grado di esercitare il feed back negativo sull’ipotalamo e sull’ipofisi; ciò causa una aumentata produzione di CRH e ACTH, i quali, a loro volta, determinano una iperstimolazione della corteccia corticosurrenalica.
Nonostante l’iperstimolazione del surrene, però, il blocco enzimatico permane ugualmente e non può essere superato; anzi, proprio in seguito alla iperstimolazione surrenalica si assiste ad un accumulo di quei precursori del cortisolo che, nella sequenza biosintetica degli ormoni surrenalici, sono posti a monte del difetto enzimatico. I precursori accumulati, pertanto, non potendo proseguire lungo la via che dovrebbe portarli verso la sintesi del cortisolo, vengono deviati verso altre vie biosintetiche che invece portano alla produzione di ormoni sessuali maschili (testosterone).
Ciò può determinare un eccesso di sintesi di ormoni sessuali e, di conseguenza, la comparsa di segni di iperandrogenismo (virilizzazione) che, nei neonati di sesso femminile, si può manifestare, già alla nascita, con ambiguità genitali mentre nei maschi si manifesta clinicamente solo in un periodo successivo, ovvero nei primi anni di vita, attraverso un aumento patologico della velocità di crescita. (Prenota una visita endocrinologica).
Contemporaneamente, alla mancata sintesi del cortisolo si può aggiungere una insufficiente produzione di aldosterone che può determinare, nei casi più gravi, un concomitante squilibrio idroelettrolitico con ipovolemia e shock.
Inoltre, alcuni studi effettuati su modelli animali (topi con deficit di 21-idrossilasi) hanno dimostrato che in questa patologia possono essere presenti anche altre alterazioni come, ad esempio, un alterato feedback ipotalamo-ipofisi-surrene e disfunzioni riguardanti non solo la corticale ma anche la midollare del surrene (sia dal punto di vista strutturale che funzionale) (3,4). La presenza di alterazioni della struttura midollare, inoltre, è stata confermata anche in alcuni esseri umani con deficit di 21-idrossilasi e ciò potrebbe spiegare la predisposizione di questi a sviluppare crisi surrenaliche acute in concomitanza di stati febbrili o eventi stressanti, anche se in terapia con una adeguata dose sostitutiva di glucocorticoidi (1,2).
{kind=link}
MODELLO ANIMALE
Sebbene il deficit di 21-idrossilasi nei topi non sia completamente paragonabile alla iperplasia surrenalica congenita dell’uomo, può essere considerato un valido modello con cui esaminare i meccanismi cellulari della patologia e su cui testare nuove possibilità terapeutiche.
Nei topi la delezione spontanea del gene della 21-idrossilasi comporta un’attività enzimatica praticamente inesistente e non è compatibile con la vita, infatti ne determina la morte all’incirca entro la prima settimana di vita.
La ghiandola surrenalica del topo affetto, inoltre, presenta una maggior espressione di mRNA della proteina regolatoria acuta della steroidogenesi (StAR) rispetto ai topi wild-type. La sintesi di questa proteina è stimolata dall’ACTH e pertanto gli elevati livelli di mRNA riscontrati sono indicativi della mancanza di feed back negativo a livello ipotalamico-ipofisario.
Inoltre uno studio ha dimostrato che, nei topi, il trattamento prenatale con desametazone (0,5-2μg/die) non sopprime adeguatamente la sintesi ormonale surrenalica, suggerendo, in tal modo, la presenza di una iperattività dell’asse corticotropo ipotalamo-ipofisi e una insensibilità del feedback negativo ai glucocorticoidi (3). Per questo motivo, infatti, è stato ipotizzato che la mancanza intrauterina di glucocorticoidi possa influenzare la sensibilità ipotalamo-ipofisaria al feedback negativo in epoca postnatale.
Il modello animale, inoltre, può fornire altre utili informazioni sulle modificazioni morfologiche a cui vanno incontro i surreni dei topi affetti da deficit di 21-idrossilasi.
Infatti se da un lato la corteccia surrenalica è decisamente allargata e mostra una marcata iperplasia delle cellule corticali, dall’altro la midollare del surrene è spesso ridotta di dimensioni e presenta una mancata migrazione delle cellule cromaffini al centro della ghiandola (3,4). Inoltre si osserva una riduzione del numero di vescicole secretorie all’interno delle cellule cromaffini e di conseguenza del contenuto di catecolammine all’interno delle stesse (4).
Un’ulteriore conferma del doppio coinvolgimento è dato dal fatto che l’espressione della feniletanolammina-N-metiltransferasi (l’enzima che converte la norepinefrina in epinefrina) è stimolata dai glucocorticoidi; pertanto proprio la ridotta sintesi di glucocorticoidi nella sindrome adrenogenitale può spiegare, probabilmente, la ridotta sintesi di catecolammine nei topi con deficit di 21-idrossilasi.
Questi dati sono confermati, nell’uomo, dal riscontro di una incompleta formazione della midollare dei surreni in 3 pazienti affetti (su 5), che hanno effettuato una surrenectomia bilaterale come terapia.
Sulla base di questi dati, quindi, sono stati effettuati degli studi sull’uomo che hanno confermato la doppia disfunzione (strutturale e funzionale) sia della corticale che della midollare del surrene. Questi ultimi dati, inoltre, assumono importanti implicazioni cliniche data la delicata situazione cardiovascolare che normalmente caratterizza questi pazienti.
Quindi anche nell’essere umano affetto dal deficit di 21-idrossilasi sono contemporaneamente coinvolti sia la corticale che la midollare del surrene e il grado di interessamento midollare può essere, addirittura, utilizzato come marker della severità della patologia (5).
Il modello animale, inoltre, si sta rivelando molto utile nel testare nuove possibilità terapeutiche come la terapia genica per la cui trattazione si rimanda al paragrafo sulle nuove prospettive terapeutiche (2,6). (Prenota una visita endocrinologica).
FENOTIPO
E’ possibile osservare un ampio spettro di fenotipi.
Innanzitutto va fatta una distinzione fra un deficit più grave, detto deficit classico, che si manifesta già in epoca neonatale o nelle prime fasi dell’infanzia con virilizzazione e insufficienza surrenalica (con o senza perdita di sali), e uno meno grave, detto deficit non classico, che può essere asintomatico o associato solo a pochi segni di iperandrogenismo e che solitamente si manifesta più tardivamente (nella fase finale dell’infanzia o addirittura in età adulta).
Nell’ambito della forma classica, inoltre, è possibile distinguere una forma più grave, detta con perdita di sale, in cui vi è un contemporaneo difetto della sintesi del cortisolo e dell’aldosterone ed una, lievemente meno grave, detta virilizzante semplice (in cui il deficit riguarda solo la sintesi del cortisolo con una sintesi di aldosterone, invece, apparentemente normale).
Nell’ambito della forma non classica, invece, è possibile distinguere una forma molto lieve (detta appunto deficit non classico) da una completamente asintomatica (detta eterozigosi) in cui l’unica differenza con la popolazione non affetta consiste negli aumentati livelli di 17-idrossi progesterone dopo stimolo con ACTH.
Il deficit classico si riscontra in circa 1 su 16000 nati, senza grosse variazioni fra le diverse popolazioni studiate.
La forma non classica, invece, è molto più frequente e si riscontra nel 0,2 % della popolazione bianca in generale anche se è di più frequente riscontro negli ebrei Ashkenazi e negli ispanici (1-2%) (1,2,7,8). (Prenota una visita endocrinologica).
Deficit classico
1) Con perdita di sale
2) Virilizzante semplice
Deficit non classico
1) Forma non classica
2) Eterozigosi
GENOTIPO
Le mutazioni del gene CYP21 sono responsabili del deficit di 21-idrossilasi.
Il gene CYP21 (CYP21A2) è situato sul cromosoma 6 nella regione altamente polimorfica del complesso di istocompatibilità HLA insieme ad uno pseudogene CYP21P (CYP21A1P). Sebbene CYP21 e il suo pseudogene abbiano il 98% della sequenza nucleotidica in comune, quest’ultimo ha accumulato innumerevoli mutazioni che rendono completamente inattivo il suo prodotto.
Fra queste ricordiamo una delezione di 8 paia di basi nell’esone 3, un frame shift nell’esone 7 ed una mutazione nonsenso nell’esone 8. Altre mutazioni dello psudogene CYP21P riguardano lo splicing dell’mRNA o la sequenza amminoacidica.
Inoltre, la posizione occupata da CYP21 all’interno della regione HLA (regione con un’alta percentuale di ricombinazione) rende questo gene molto suscettibile a cambiamenti di sequenza nucleotidica. Per questo motivo il gene CYP21 è un gene estremamente polimorfo ed estremamente variabile, per ciò che riguarda la sequenza, da individuo ad individuo.
Una ulteriore conferma di ciò è la dimostrazione che anche negli spermatozooi le ricombinazioni spontanee fra CYP21 e CYP21P sono molto frequenti e anche qui si tratta, nella maggior parte dei casi, di conversioni geniche o delezioni.
Sostanzialmente, però, si può affermare che il deficit di 21-idrossilasi è prevalentemente (ma non solo) una conseguenza del trasferimento di sequenze nucleotidiche dallo pseudogene al gene CYP21. Il passaggio di sequenze nucleotidiche normalmente presenti nello pseudogene inattivo all’interno del gene attivo rendono quest’ultimo incapace di codificare per un enzima funzionante. Il trasferimento di sequenze fra geni omologhi è solitamente detto “conversione genica” ma rimane un fenomeno ancora non perfettamente chiarito.
Infatti, circa il 75% dei deficit di 21-idrossilasi è dovuto a mutazioni (delezioni) presenti nello pseudogene che vengono trasferite nel gene funzionante durante la mitosi attraverso il meccanismo di “conversione genica”. (Prenota un test genetico).
Circa il 20% sono ricombinazioni meiotiche che comportano la perdita di un segmento genico di circa 30 kbasi che comprende l’estremità 3’-terminale dello pseudogene, la sequenza del gene del fattore C4B del complemento e l’estremita 5’-iniziale di CYP21, producendo uno pseudogene chimerico non funzionante.
Il restante 5% dei casi, invece, può essere causato da oltre 60 mutazioni diverse fin ora identificate (1).
Va ricordato, infine, che circa l’1-2% degli alleli affetti sono il frutto di mutazioni spontanee e non sono ereditati dai genitori; pertanto è sempre bene accertarsi del reale genotipo dei genitori per escludere la possibilità di una mutazione de novo (9,10).
CORRELAZIONE GENOTIPO-FENOTIPO
La correlazione genotipo-fenotipo è stata studiata in diverse popolazioni tenendo conto delle varie differenze fra etnie e razze.
Solitamente a varie, specifiche mutazioni corrispondono solo tre differenti fenotipi di deficit di 21-idrossilasi. Infatti le mutazioni di CYP21 possono essere classificate in tre categorie a seconda del livello di attività residua dell’enzima 21-idrossilasi.
Il primo gruppo è rappresentato da quelle mutazioni, come delezioni o mutazioni nonsenso, che annullano completamente l’attività dell’enzima. Questo tipo di mutazioni sono spesso associate con la forma clinica più grave (con perdita di sale).
Il secondo gruppo di mutazioni è rappresentato prevalentemente dalla mutazione missenso Ile172Asn (I172N) che porta alla sintesi di un enzima con una attività catalitica pari all’1-2% di quella normale. Questo tipo di mutazione, però, comporta una sintesi pressoché normale di aldosterone e pertanto si riscontra soprattutto nella forma clinica di deficit virilizzante semplice.
Il terzo gruppo comprende quelle mutazioni come Val281Leu (V281L) e Pro30Leu (P30L) che portano ad un enzima con un’attività residua compresa fra il 20 e il 60 % della normale attività enzimatica; questo tipo di mutazioni si riscontrano solitamente nei pazienti affetti da deficit non classico di 21-idrossilasi (Figura 4).
L’eterozigosi per due differenti mutazioni di CYP21, invece, porta solitamente ad un fenotipo determinato dall’allele affetto meno severamente.
Quando il fenotipo del deficit di 21-idrossilasi viene quantificato attraverso i livelli di 17-idrossi progesterone o attraverso degli scores indicanti il grado di iperandrogenismo o di perdita salina, la variazione allelica nel genotipo in CYP21 spiega l’80-90% della variazione fenotipica. Con questo si vuole dire che non sempre ad uno specifico genotipo corrisponde un altrettanto specifico fenotipo. Questa discrepanza fra genotipo-fenotipo può essere spiegata dalla diversa sensibilità periferica agli androgeni o dalla presenza di altri geni che possono influenzare la steroidogenesi surrenalica.
Va ricordato, infatti, che sebbene il deficit di 21-idrossilasi sia un disordine monogenico, il background genetico del soggetto potrà influenzare l’espressione del gene CYP21 esattamente come avviene in altre patologie umane. Tra questi geni potrebbero avere un ruolo importante quelli che regolano la produzione e l’attività periferica di androgeni ed estrogeni e quelli coinvolti nella ritenzione del sodio.
Un’altra causa di variabilità fenotipica è la cosiddetta perdita delle variazioni di splicing. Infatti una mutazione nel secondo introne, trasferita dal gene CYP21P attraverso il meccanismo di conversione genica (sostituzione di una guanina in posizione 656 con una adenina), determina uno splicing anomalo dell’mRNA ed è responsabile di circa il 25% dei deficit classici di 21-idrossilasi. Altri dati, però, sembrano non confermare questa tesi perché hanno evidenziato che una piccola quantità di mRNA viene comunque sottoposta ad un normale processo di splicing.
Va ricordato, infine, che una attività enzimatica residua del solo 1-2% del normale può essere sufficiente, da sola, a modificare il fenotipo del paziente favorendo la forma clinica meno grave (virilizzante semplice) al posto di quella più grave (con perdita di sale) (2,11,12). (Prenota una visita endocrinologica).
CLINICA
DEFICIT CLASSICO :
1) Perdita di sale
Circa il 75% dei pazienti con deficit classico di 21-idrossilasi è incapace di sintetizzare adeguate quantità di aldosterone; inoltre i precursori accumulati (progesterone e 17-idrossi progesterone) agiscono anche come antagonisti recettoriali dei mineralcorticoidi e possono ulteriormente peggiorare la sintomatologia dovuta alla mancanza di aldosterone.
L’aldosterone è l’ormone che regola l’omeostasi del sodio e pertanto una sua mancanza determina, nei pazienti non trattati, una aumentata escrezione di sodio con conseguente iponatremia, ipovolemia e iperreninemia. Inoltre, siccome il potassio a livello renale viene scambiato con il sodio, l’elevata escrezione di quest’ultimo comporta un accumulo di potassio con conseguente iperkaliemia, soprattutto durante l’infanzia.
In questi pazienti il deficit di cortisolo complica ulteriormente il quadro clinico determinando un peggioramento della funzione cardiaca, una riduzione della risposta vascolare alle catecolammine ed una riduzione della filtrazione glomerulare.
Infatti, dato che la midollare del surrene è in parte dipendente dai glucocorticoidi, nei pazienti con perdita di sale si può avere anche un deficit di catecolammine che può peggiorare la condizione di shock.
Pertanto, la contemporanea assenza di cortisolo e aldosterone causa frequentemente disidratazione iponatriemica e, nei casi non trattati, anche shock ipovolemico. Clinicamente la perdita di sale si manifesta tipicamente fra il settimo e il quattordicesimo giorno di vita con vomito, perdita di peso, letargia, iponatremia e iperkaliemia.
I pazienti con il deficit classico di 21-idrossilasi con perdita di sale vengono identificati attraverso il dosaggio degli elettroliti, della renina e dell’aldosterone che mostrano, iperreninemia con bassi valori di aldosterone.
La valutazione della renina va fatta utilizzando i range di riferimento per età, dato che l’attività plasmatica reninica è normalmente più elevata nei neonati rispetto ai bambini un pò più grandi (1).
Nella forma con perdita di sale, oltre ai sintomi ed ai segni precedentemente descritti si riscontrano anche tutte le conseguenze dell’iperandrogenismo descritte nella forma virilizzante semplice. (Prenota una visita endocrinologica).
2)Virilizzante semplice
Questo genere di paziente non è in grado di produrre sufficienti quantità di cortisolo ma è perfettamente in grado di produrre adeguate quantità di aldosterone e quindi di mantenere un corretto bilancio elettrolitico.
Il problema, però, è che se nelle femmine la diagnosi è generalmente posta alla nascita per la presenza di ambiguità genitali (pseudoermafroditismo), nel maschio affetto la diagnosi può essere ritardata anche di molti anni. Solitamente, infatti, nel maschio la diagnosi viene effettuata per lo sviluppo di pubertà precoce con comparsa di pubarca precoce o per la comparsa di una improvvisa accelerazione della velocità di crescita, in un’età generalmente compresa fra i 2 e i 4 anni.
Per cui se non viene effettuato uno screening neonatale la diagnosi di deficit di 21-idrossilasi, nel maschio, può essere effettuata solo quando compaiono i segni dell’iperandrogenismo e, purtroppo, un ritardo nella diagnosi si associa non solamente ad una minor efficacia della terapia farmacologica ma anche alla comparsa di pubertà precoce e quindi, spesso, ad una condizione finale di bassa statura patologica (1). (Prenota una visita auxologica).
I segni ed i sintomi clinici che più frequentemente si riscontrano nel deficit classico sono i seguenti:
-Ambiguità dei genitali:
Le femmine con un deficit classico di 21-idrossilasi sono esposte, a partire dalla settima settimana di gestazione, ad elevate concentrazioni di androgeni.
Questa sovraesposizione agli androgeni porta ad un grado variabile di virilizzazione del feto che, pertanto, alla nascita può avere delle ambiguità genitali tali da causare, addirittura, difficoltà nell’assegnazione del sesso fenotipico (pseudoermafroditismo); solitamente le ambiguità genitali più frequenti sono: clitoridomegalia, grandi labbra rugose e parzialmente fuse ed un seno urogenitale comune (invece di uretra e vagina separate). (Prenota un’ecografia pelvica).
Solitamente, però, l’utero, le tube di falloppio e l’ovaio hanno una conformazione normale. La peculiarità e l’evidenza dei segni di virilizzazione, pertanto, consente, nelle donne, una diagnosi del deficit classico di 21-idrossilasi già al momento della nascita.
Al contrario, invece, i maschi affetti, alla nascita, non presentano, ovviamente, alcun segno genitale della patologia ad eccezione di una variabile, e comunque lieve, iperpigmentazione dello scroto ed un certo ingrandimento del pene. Pertanto nei maschi la diagnosi della patologia alla nascita viene spesso misconosciuta a meno che non vi sia un concomitante deficit di aldosterone, che determina forti crisi ipotensive e perdita di sale, o non venga effettuato un adeguato screening neonatale (1).
-Virilizzazione postanale:
Nei pazienti non trattati, o inadeguatamente trattati, l’esposizione per lungo tempo ad un eccesso di androgeni determina una rapida crescita corporea (effetto dovuto prevalentemente agli androgeni) ed un avanzamento dell’età ossea con una precoce chiusura delle cartilagini epifisarie (effetto dovuto prevalentemente agli estrogeni formatisi dalla aromatizzazione periferica degli androgeni).
Anche il pubarca ed il telarca possono comparire precocemente.
Con il passare del tempo le dimensioni del clitoride possono continuare ad aumentare nelle femmine così come quelle del pene nel maschio, anche se il volume dei testicoli rimane sempre piccolo (1). L’iperandrogenismo, inoltre, sia nella forma classica che in quella non classica, può essere una condizione predisponente allo sviluppo della sindrome dell’ovaio policistico, come dimostrato in uno studio su femmine transessuali. Secondo questo studio, infatti, oltre il 70% delle donne transessuali ha sviluppato una sindrome dell’ovaio policistico secondaria all’iperandrogenismo iatrogeno (13). Pertanto sarebbe opportuno instaurare precocemente una adeguata terapia per prevenire il deterioramento e la sclerosi ovarica progressiva che si riscontra nella sindrome adreno-genitale e, appunto, nella sindrome dell’ovaio policistico. Questo processo, inoltre, è ulteriormente accelerato dalla eventuale compresenza di insulino-resistenza (14). Infatti circa il 25% delle donne con iperplasia surrenalica congenita hanno la sindrome dell’ovaio policistico associata con la sindrome X (14). (Prenota una visita endocrinologica).
Infine va ricordato che la lunga esposizione ad elevati livelli di androgeni può determinare l’attivazione dell’asse ipotalamo-ipofisi-gonadi determinando una pubertà precoce vera.
–Bassa statura patologica:
Una meta-analisi effettuata sui dati ottenuti da 18 centri diversi ha evidenziato che l’altezza finale, nei pazienti con la forma classica di iperplasia surrenalica congenita, è all’incirca 1,4 SD al disotto della popolazione normale (15). (Prenota una visita auxologica).
Inoltre va ricordato che sia i pazienti sotto-trattati che quelli iper-trattati sono a rischio di bassa statura. Nel primo caso la bassa statura è conseguente alla precoce chiusura delle cartilagini epifisarie indotta dagli alti livelli di ormoni sessuali; nel secondo caso, invece, la bassa statura è una conseguenza dell’inibizione dell’asse della crescita indotta dalla elevata dose di glucocorticoidi introdotti con la terapia (1,16,17).
-Disfunzioni della riproduzione:
Nelle donne con qualsiasi forma di deficit di 21-idrossilasi possono comparire oligomenorrea o amenorrea, soprattutto durante il periodo adolescenziale.
Inoltre, l’esposizione ad elevati livelli di androgeni durante il periodo prenatale può influenzare il successivo comportamento sessuale. Infatti alcuni studi hanno evidenziato che alcune femmine affette, durante l’infanzia, potevano avere un comportamento più tipicamente maschile per ciò che riguarda i giochi preferiti, il ruolo assunto negli stessi e l’aggressività nel comportamento (18). Nonostante ciò, comunque, la maggior parte delle donne sono comunque eterosessuali e la loro identità sessuale è invariabilmente di tipo femminile.
Inoltre, sebbene la fertilità possa essere notevolmente ridotta, nel caso in cui la produzione di androgeni surrenalici non sia adeguatamente soppressa con la terapia steroidea, molte donne con deficit di 21-idrossilasi sono ugualmente in grado di concepire e di portare a termine con successo una gravidanza.
Si può pertanto affermare che circa l’80% delle donne con deficit virilizzante semplice ed approssimativamente il 60% di quelle con perdita di sale sono fertili (19).
Confrontati con le donne affette, invece, gli uomini con deficit di 21-idrossilasi hanno molti meno problemi della funzione riproduttiva. Molti di essi, infatti, hanno uno spermiogramma nella norma ed una normale capacità riproduttiva.
Tuttavia, nei maschi affetti una forma relativamente frequente di anormalità gonadica è lo sviluppo di tessuto surrenalico ectopico, il più delle volte a livello testicolare e quasi sempre bilaterale (testicular adrenal rest tissue) (20). (Prenota una visita andrologica).
Il tessuto surrenalico ectopico è, dal punto di vista biochimico, del tutto identico a quello del corticosurrene e la sua funzionalità, esattamente come il tessuto surrenalico, può essere stimolata dall’ACTH o soppressa dalla terapia con glicocorticoidi (21). Addirittura, in alcuni pazienti affetti da iperplasia surrenalica congenita la virilizzazione persistente è stata attribuita proprio al tessuto surrenalico testicolare attivato (22).
Embriologicamente parlando, sia le gonadi che i surreni originano dal mesoderma del tratto urogenitale e proprio per questo motivo il riscontro di tessuto surrenalico (adrenal remnants) è molto frequente nelle gonadi, soprattutto nel testicolo, sebbene possa riscontrarsi anche in altre parti del corpo (plesso celiaco, legamento largo e ovaie) (20).
Questo è un tessuto definito di tipo pseudotumorale e può essere riscontrato attraverso un esame ecografico, ancora prima che diventi clinicamente palpabile. In uno studio NIH, infatti, l’ecografia testicolare ha permesso l’individuazione del testicular adrenal rest tissue nel 30% dei pazienti con deficit classico indagati e di questi solo il 5% aveva una massa clinicamente palabile (23). Pertanto, onde evitare che tale reperto sfugga alla diagnosi è necessario sottoporre il paziente affetto ad approfondimenti diagnostici di tipo strumentale.
L’esame ultrasonografico mostra delle masse bilaterali ed ipoecogene con altri aspetti caratteristici ma non patognomonici del testicular adrenal rest tissue. La bilateralità della lesione, però, permette la diagnosi differenziale con le altre masse testicolari (compresi i tumori) che tendono ad essere quasi sempre monolaterali. Inoltre il color Doppler può mostrare un flusso ematico ordinato e non turbolento all’interno della lesione, mentre nel caso dei carcinomi testicolari la vascolarizzazione è più spesso disordinata e irregolare (20).
Alla risonanza magnetica nucleare (RMN), invece, il comportamento è simile a quello della ghiandola surrenalica normale (24); la maggior parte delle masse, infatti, sono isointense nelle immagini T1-pesate, ipointense nelle immagini T2-pesate e presentano un diffuso enhancement dopo mezzo di contrasto.
In ogni modo data la maggior praticità ed il minor costo dell’esame ultrasonografico, proprio quest’ultima dovrebbe essere considerata la metodica di elezione nella diagnosi e nel follow up del testicular adrenal rest tissue (Figura 6).
Il tessuto surrenalico ectopico, in alcuni casi, può degenerare e subire una trasformazione di tipo neoplastico; nella maggior parte dei casi, comunque, si tratta di un tumore benigno, ma per averne la certezza è necessario un approfondimento diagnostico mediante l’esecuzione di una biopsia testicolare o, eventualmente, una orchiectomia parziale.
Nei maschi con perdita di sale lo sviluppo di tessuto surrenalico nel testicolo può determinare oligo-azoospermia e quindi sterilità (25,38).
Nel caso di testicular adrenal rest tissue di picole dimensioni la terapia consiste nella soppressione dell’asse ipotalamo-ipofisi-surrene (HPA) con il dosaggio standard di glucocorticoidi (desametazone). Se la lesione è di grosse dimensioni e il paziente è sintomatico o infertile è necessaria una terapia con glucocorticoidi ad alto dosaggio, ma vanno sempre considerati gli effetti collaterali di questo tipo di trattamento.
Nel caso in cui la massa sia irresponsiva alla terapia steroidea o addirittura cresca di dimensioni sarebbe opportuno effettuare ulteriori accertamenti diagnostici (biopsia testicolare) (38).
L’orchiectomia, pur essendo il trattamento d’elezione nelle neoplasie testicolari, è raramente indicata come terapia del testicular adrenal rest tissue; infatti, indicazioni all’orchiectomia sono solamente il sospetto di malignità o la persistenza del dolore (1).
DEFICIT NON CLASSICO
1)Forma non classica
I pazienti con un deficit non classico di 21-idrossilasi riescono a sintetizzare adeguate quantità di cortisolo e aldosterone; questo risultato, però, può essere raggiunto solo con una iperstimolazione del surrene e quindi a spese di un’aumentata sintesi di precursori degli ormoni sessuali.
Solitamente la maggior parte dei deficit non classici non viene diagnosticata con screening neonatale per il riscontro di valori di 17-idrossi progesterone relativamente bassi e pertanto solo una piccola parte viene effettivamente scoperta alla nascita.
Non è ancora nota quale percentuale di casi diventerà poi sintomatica, ma alcuni studi sembrano dimostrare che, nella maggior parte dei bambini con deficit non classico, non compariranno mai gravi segni di iperandrogenismo e, solo in una minoranza dei casi, invece, si possono riscontrare disturbi più gravi come l’accelerazione patologica della velocità di crescita, l’eccessivo avanzamento dell’età ossea o la comparsa di pubertà precoce. (Prenota una visita endocrinologica).
Ovviamente la sintomatologia del deficit non classico di 21-idrossilasi varia a seconda del sesso. Pertanto, nelle donne con deficit non classico prevalgono i segni clinici legati ad una condizione di iperandrogenismo moderato-lieve; l’irsutismo rappresenta il sintomo di presentazione più frequente nelle donne sintomatiche (60%) seguito dall’oligomenorrea (54%) e dall’acne (33%). In alcuni casi può essere presente infertilità.
Teoricamente, inoltre, nelle donne dovrebbe essere presente anche un aumentato rischio di neoplasie dell’apparato riproduttivo conseguente all’anovulazione cronica non opposta dall’azione estrogenica ma questo dato non è stato ancora dimostrato.
La riduzione della fertilità è un’indicazione al trattamento con glucocorticoidi, nelle femmine come nei maschi, anche se la frequenza del deficit non classico all’interno della popolazione infertile non è differente da quella della popolazione generale; va aggiunto, inoltre, che il sintomo infertilità è presente solo nel 13% delle donne con deficit non classico di 21-idrossilasi.
Anche nell’ambito dell’irsutismo, l’iperplasia surrenalica congenita rappresenta solo il 5-10% del totale, così come fra le cause di adrenarca precoce l’iperplasia surrenalica congenita rappresenta solo il 5-10%.
Come si può notare, queste manifestazioni cliniche del deficit non classico di 21-idrossilasi sono molto simili a quelle che si riscontrano nella policistosi ovarica tanto che, se è presente una concomitante insulino-resistenza, spesso le due patologie possono anche fondersi e confondersi fra loro. Va ricordato, infatti, come già detto in precedenza, che l’iperandrogenismo predispone allo sviluppo della sindrome dell’ovaio policistico (13,14). (Prenota una visita endocrinologica).
Secondo alcuni studi recenti, inoltre, l’iperplasia surrenalica congenita potrebbe essere associata ad una aumentata incidenza di adenomi surenalici o di incidentalomi (26). Infatti, sia nel deficit classico che nel non-classico, è stato dimostrato un aumentato riscontro di adenomi corticosurrenalici alla risonanza magnetica (26).
I maschi con deficit non classico, invece, sono spesso asintomatici ma possono occasionalmente presentare pubertà precoce o lo sviluppo di tessuto surrenalico testicolare che, come nella forma classica, può determinare un senso di peso, a volte dolore e nei casi più gravi oligospermia, azoospermia e infertilità (1). (Prenota una visita andrologica).
2)Eterozigosi
I pazienti che sono eterozigoti per la mutazione del gene CYP21 hanno spesso valori di 17-idrossi progesterone basale per lo più sovrapponibili a quelli riscontrabili nei soggetti normali, mentre dopo lo stimolo con ACTH sono solitamente più elevati rispetto a quelli della popolazione non affetta ma non hanno segni e/o sintomi della patologia.
Sebbene sia stato suggerito che i soggetti eterozigoti potrebbero avere con maggior probabilità dei segni di iperandrogenismo rispetto alla popolazione non affetta, questo dato non è stato confermato da studi più recenti (1).
DIAGNOSI
Il deficit classico di 21-idrossilasi è caratterizzato da livelli marcatamente elevati di 17-idrossi progesterone, il principale substrato dell’enzima.
Nei bambini normali i livelli basali di 17-idrossi progesterone sono inferiori ai 100 ng/dl (3 nmol/l) mentre nei bambini affetti dalla forma classica i valori del substrato in questione superano i 10000 ng/dl (300 nmol/l); in realtà non vi è univocità sul valore di 17-idrossi progesterone diagnostico per deficit classico; infatti sono stati proposti diversi valori diagnostici ognuno dei quali si associa, a seconda dello studio effettuato, ad una diversa sensibilità ed una diversa specificità.
In ogni modo, questa differenza di valori rende possibile uno screening nei neonati riducendo, così, il ritardo nella diagnosi, soprattutto nei maschi, e diminuendo la morbilità e la mortalità delle crisi di insufficienza surrenalica.
Solitamente i neonati con perdita di sale hanno livelli basali di 17-idrossi progesterone più elevati di quelli dei bambini con il deficit virilizzante semplice; inoltre il riscontro di valori basali molto elevati di 17-idrossi progesterone sono già diagnostici per un deficit classico e non richiedono un test di stimolo con ACTH.
Circa il 10 % dei nati a termine affetti da una forma severa della patologia hanno valori basali di 17-idrossi progesterone normale-bassi. Falsi negativi, inoltre, si ottengono soprattutto nei neonati che sono dimessi precocemente dall’ospedale in cui lo screening è stato effettuato prima del terzo giorno di vita evidenziando valori di 17-idrossi progesterone falsamente bassi.
Al contrario si possono riscontrare valori falsamente elevati di 17-idrossi progesterone in neonati malati o nei nati prematuri (soprattutto se nati prima della 31º settimana) in cui, in realtà, non vi è nessun difetto nella sintesi degli steroidi surrenalici.
Per evitare che nessun bambino con il deficit di 21-idrossilasi sfugga alla diagnosi con lo screening basale bisognerebbe abbassare i valori patologici di 17-idrossi progesterone a livelli così bassi che aumenterebbero di molto i falsi positivi e si otterrebbe un valore predittivo dello screening del solo 2%.
Pertanto per aumentare l’accuratezza diagnostica dello screening sono state tentate diverse soluzioni tra cui quella di basare i livelli di riferimento del 17-idrossi progesterone al peso e all’età gestazionale, e quella di commisurare i valori di 17-idrossi progesterone a quelli del cortisolo plasmatico.
Il modo migliore per differenziare un deficit di 21-idrossilasi dagli altri deficit enzimatici, però, rimane il test di stimolo con ACTH (effettuato iniettando 250 μg di corticotropina in bolo e.v.) e misurando i valori di 17-idrossi progesterone, basali e 60 minuti dopo lo stimolo.
Tranne che per i bambini prematuri non ci sono differenze nei criteri di diagnosi del deficit di 21-idrossilasi sulla base dei valori di 17-idrossi progesterone.
Le differenze nei valori di 17-idrossi progesterone, solitamente, riflettono una diversa gravità del difetto enzimatico.
I pazienti con perdita di sale hanno i valori più elevati di 17-idrossi progesterone dopo stimolo con corticotropina (superiori a 100000 ng/dl (3000 nmol/l)) seguiti da quelli con la forma virilizzante semplice (con valori compresi fra 10000 e 30000 ng/dl (300-1000 nmol/l)). (Prenota una visita endocrinologica).
Invece, studi effettuati su pazienti affetti da deficit non classico hanno dimostrato che i valori di 17-idrossi progesterone seguono un ritmo circadiano (con un picco mattutino) e sono, nella maggior parte dei casi, nel range di normalità. Pertanto il dosaggio casuale del 17-idrossi progesterone basale può risultare non diagnostico nei soggetti con deficit non classico, a meno che i valori non siano ottenuti con un prelievo effettuato il mattino presto. Va ricordato, però, che anche il 17-idrossi progesterone mattutino risulta spesso nella norma e va usato solo nello screening della patologia non classica perché ha una sensibilità ed una specificità diagnostica molto inferiore a quella del test di stimolo con ACTH.
La certezza diagnostica, infatti, si ottiene solo attraverso l’esecuzione del test di stimolo con ACTH ed è necessario che il 17-idrossi progesterone (dopo lo stimolo con ACTH) sia almeno superiore a 45,5 nmol/l. In realtà anche nel caso di deficit non classico sono stati proposti diversi valori cut off ad ognuno dei quali si associa, a seconda dello studio effettuato, ad una diversa sensibilità e ad una diversa specificità diagnostica.
In linea di massima, rispetto al deficit classico, i pazienti con la forma non classica hanno valori di 17-idrossi progesterone dopo stimolo meno elevati (fra 1500 e 10000 ng/dl (50-300 nmol/l)); valori ancora più bassi sono riscontrabili nei soggetti eterozigoti (fra 300 e 1500 ng/dl (<50 nmol/l)) mentre nei soggetti normali i valori di 17-idrossi progesterone sono solitamente inferiori a 300 ng/dl. (Prenota una visita endocrinologica).
Va ricordato, però, che nel deficit di 21-idrossilasi possono risultare elevati anche altri ormoni come il progesterone, l’androstenedione e in quantità minore il testosterone. Anche il 21-desossicortisolo risulta solitamente elevato ma il suo dosaggio non è routinario.
L’analisi genetica della mutazione, infine, conferma la diagnosi ed è usata nei programmi di screening nei neonati (1).
TERAPIA
Glucocorticoidi
Nel deficit classico di 21-idrossilasi la terapia prevede un trattamento a lungo termine con glucorticoidi per inibire l’eccessiva secrezione di CRH e ACTH da parte dell’ipotalamo e dell’ipofisi, rispettivamente, e di conseguenza per ridurre l’eccessiva produzione di ormoni sessuali surrenalici.
Nei bambini il farmaco da preferire è l’idrocortisone somministrato ad un dosaggio di 10-20 mg per metro quadro di superficie corporea al giorno frazionata in tre dosi. Questo dosaggio è comunque lievemente superiore alla secrezione fisiologica di cortisolo.
Per i conosciuti effetti collaterali dei glucorticoidi si sconsiglia un dosaggio superiore a 25 mg per metro quadro.
Dosi superiori a 100 mg per metro quadro, invece, vanno somministrate durante gli episodi di insufficienza surrenalica acuta o comunque nelle situazioni a rischio di vita.
Il monitoraggio della terapia va monitorato attraverso il dosaggio dei livelli di 17-idrossi progesterone e di androstenedione e durante la terapia i bambini vanno sottoposti a periodici controlli medici per valutarne annualmente l’età ossea e l’andamento della crescita. (Prenota una visita endocrinologica).
Il goal terapeutico sarebbe utilizzare il dosaggio terapeutico minimo sufficiente a sopprimere la produzione degli androgeni surrenalici e contemporaneamente a mantenere un accrescimento corporeo ed un peso adeguato.
L’obbiettivo non deve essere raggiungere valori normali di 17-idrossi progesterone, perché per raggiungere tale risultato sarebbero necessari dosaggi sovrafisiologici di idrocortisone tali da causare, a volte, addirittura una sindrome di Cushing iatrogena (27). Piuttosto i livelli di 17-idrossi progesterone devono essere solo parzialmente soppressi a valori compresi fra 100 e 1000 ng/dl (3-30 nmol/l).
I livelli di testosterone (il cui dosaggio è utile solo nei pazienti prepuberi) e androstenedione, invece, devono essere mantenuti entro i livelli di riferimento per sesso ed età; per realizzare ciò, però, è spesso necessario aumentare il dosaggio dei glucocorticoidi.
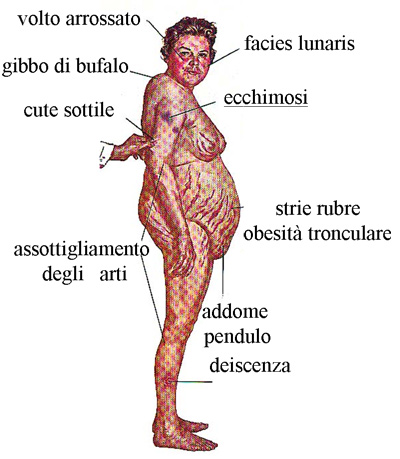
La difficoltà maggiore nella terapia del deficit classico di 21-idrossilasi consiste nel trovare il giusto dosaggio farmacologico che impedisca l’iperandrogenismo senza causare, al tempo stesso, una condizione di ipercortisolismo; l’ipercortisolismo iatrogeno si rende clinicamente evidente per la comparsa di incremento ponderale, ridistribuzione tronculare dell’adipe, intolleranza glucidica, ipertensione e dislipidemia (Figura 7).
Nei bambini si preferisce utilizzare l’idrocortisone per la sua farmacocinetica. Infatti il suo breve tempo di dimezzamento riduce al minimo il rallentamento della crescita e con esso tutti gli altri effetti collaterali dei glucocorticoidi più potenti ed a maggiore durata d’azione (prednisone e desametazone). D’altra parte, però, proprio il suo breve tempo di dimezzamento rende un’unica somministrazione giornaliera inefficace nel sopprimere adeguatamente la stimolazione surrenalica.
Il discorso è diverso, invece, per gli adolescenti e gli adulti; questi, infatti, devono essere trattati con prednisone (ad un dosaggio variabile fra 5 e 7,5 mg/die frazionato in due dosi al giorno) o con desametazone (ad un dosaggio variabile fra 0,25 e 0,5 mg/die somministrato in una o due dosi al giorno).
Questi pazienti devono essere strettamente monitorizzati per valutare la comparsa di segni di ipercortisolismo come strie rubre, osteopenia, ipertensione o rapido incremento ponderale.
I maschi con testicular adrenal rest tissue, spesso, necessitano di un dosaggio di desametazone maggiore per sopprimere i livelli di ACTH. In ogni caso la terapia soppressiva nel maschio va effettuata anche in età adulta per ridurre il rischio di sviluppo di testicular adrenal rest tissue.
Nel deficit non classico di 21-idrossilasi, invece, il trattamento farmacologico con glucocorticoidi non è indicato nei pazienti asintomatici, visto che gli effetti collaterali dei glucocorticoidi finirebbero per superarne i benefici.
I maschi con deficit non classico di 21-idrossilasi, solitamente, non richiedono alcun trattamento. Il trattamento con glicocorticoidi, invece, potrebbe essere indicato nei bambini maschi con pubertà precoce, con un aumento della velocità di crescita e con un avanzamento dell’età ossea, per ridurre il rischio di ipostaturismo. Ancora più raramente la terapia steroidea potrebbe essere indicata, nei maschi adulti, per impedire lo sviluppo di tessuto surrenalico residuo nel testicolo.
Invece nelle femmine, soprattutto nelle adolescenti e nelle giovani donne con segni di virilizzazione, può essere preso in considerazione anche un trattamento farmacologico alternativo, come l’utilizzo della pillola anticoncezionale estro-progestinca, di un farmaco con effetto antiandrogeno o di entrambi.
Lo spironolattone e la flutamide, il cui utilizzo non è formalmente approvato per questa patologia, sono, di fatto, gli antiandrogeni più utilizzati.
La pillola anticoncezionale, invece, impedendo la sclerotizzazione e lo sviluppo di cisti ovariche, riduce, quindi, anche la sintesi degli androgeni. Inoltre, ostacolando la gravidanza, impedisce la nascita di bambini maschi femminilizzati, in donne che stanno assumendo una terapia antiandrogena. Chiaramente è preferibile un preparato estroprogestinico che abbia una componente progestinica con scarse proprietà androgeniche o addirittura con proprietà antiandrogene (ciproterone).
Nel caso in cui si desideri una gravidanza, invece, può esser necessaria una terapia con glucocorticoidi; in tal caso la terapia combinata con estroprogestinici e antiandrogeni deve essere ovviamente sospesa.
Mineralcorticoidi
I bambini con il deficit classico di 21-idrossilasi con perdita di sale oltre alla terapia con glucocorticoidi richiedono un supplemento terapeutico con mineralcorticoidi (solitamente fludrocortisone 0,1-0,2 mg/die) e cloruro di sodio (NaCl 1-2 g o 17-34 mmol/die). A differenza di quanto avviene per i glucocorticoidi il dosaggio terapeutico del fludrocortisone non è dipendente dal peso corporeo e pertanto non presenta sostanziali differenze fra il bambino e l’adulto.
Il sodio contenuto nel latte materno è solitamente insufficiente a compensare l’eventuale perdita di sodio e pertanto è necessaria una terapia con NaCl; i bambini più grandi, invece, non richiedono necessariamente un supplemento di NaCl, anzi, spesso si rende necessaria anche una riduzione del dosaggio del fludrocortisone stesso.
I pazienti con la forma virilizzante semplice non necessitano, per definizione, dei mineralcorticoidi ma, in alcuni casi, possono essere trattati con fludrocortisone perché in questo modo si raggiunge prima una maggior soppressione dell’asse surrenalico e di conseguenza si riduce il dosaggio di glucorticoidi richiesto per ottenere degli adeguati livelli di 17-idrossi progesterone.
Per monitorare l’adeguatezza della terapia sostitutiva con mineralcorticoidi e cloruro di sodio bisogna valutare l’attività plasmatica reninica o direttamente la renina dosata con metodica RIA. La presenza di ipotensione, iperkaliemia ed elevati livelli di renina suggeriscono un aumento del dosaggio; al contrario l’ipertensione, l’edema, la tachicardia e i valori soppressi di renina indicano un sovradosaggio dei mineralcorticoidi. (Prenota una visita endocrinologica).
Gli aggiustamenti del dosaggio vanno effettuati nell’ordine di 0,05-0,1 mg.
Va segnalato, infine, che un sovradosaggio del fludrocortisone può determinare un ritardo nella crescita (1).
Trattamento delle ambiguità genitali
Nel passato la correzione chirurgica delle ambiguità genitali veniva effettuata con un solo intervento, eseguito solitamente tra i 2 e i 6 mesi di vita, epoca in cui i tessuti sono particolarmente malleabili e il trauma psicologico legato all’intervento è ridotto al minimo. Gli studi retrospettivi effettuati su questo genere di interventi, però, hanno evidenziato un outcome non soddisfacente, sia dal punto di vista funzionale che estetico; mentre sulle nuove procedure chirurgiche non sono stati ancora effettuati studi che ne valutino l’outcome a lungo termine. Bisogna tener presente, però, che spesso l’intervento effettuato in età adolescenziale può avere delle ripercussioni psicologiche non indifferenti e attualmente si preferisce affiancare una psicoterapia di supporto che coinvolga il paziente e tutta la sua famiglia.
Inoltre sarebbe opportuno che l’intervento chirurgico fosse effettuato da chirurghi con una buona esperienza in merito.
Infine, soprattutto durante il passaggio dalla fase adolescenziale all’età adulta è consigliabile approcciare il paziente in modo multidisciplinare coinvolgendo un team di specialisti (ginecologo, endocrinologo, psicologo etc..).
DIAGNOSI E TERAPIA PRENATALE
Lo studio genetico prenatale è effettuato in tutte le famiglie affette e ciò ha aperto la controversia sull’opportunità di effettuare un trattamento prenatale.
La somministrazione di desametazone alla madre riduce le ambiguità genitali nelle femmine affette da deficit di 21-idrossilasi perché presumibilmente riduce la sintesi surrenalica di ormoni sessuali maschili impedendo così la virilizzazione del feto femmina. Il trattamento steroideo, quindi, va effettuato solo nel caso in cui il feto sia di sesso femminile.
Viene utilizzato il desametazone perché è un composto che non viene inattivato dall’enzima placentare 11β-idrossisteroido-deidrogenasi.
Il dosaggio del farmaco è di 20 μg per Kg di peso corporeo al giorno (calcolato in base al peso corporeo della mamma prima dell’inizio della gravidanza) frazionato in tre dosi. Per prevenire la virilizzazione del feto è necessario somministrare il farmaco precocemente nel primo trimestre in tutte le donne i cui feti sono a rischio di deficit di 21-idrossilasi.
Un eventuale insuccesso della terapia, quindi, può essere una conseguenza della inopportuna sospensione della terapia durante la gravidanza, dell’impiego di un dosaggio farmacologico insufficiente o della scarsa compliance alla terapia. Altri studi, però, sembrano non dare particolare importanza a questi aspetti in quanto l’eventuale scarso successo terapeutico potrebbe essere solo una conseguenza dell’estrema variabilità fenotipica della malattia; infatti si possono osservare diversi gradi di virilizzazione anche fra le bambine affette, appartenenti alla stessa famiglia, a prescindere dall’aver effettuato o meno la terapia prenatale.
Va ricordato, inoltre, che il deficit di 21-idrossilasi è una patologia autosomica recessiva e pertanto, la probabilità che da due genitori portatori possa nascere una femmina affetta è solo di 1 su 8. Questo, chiaramente, vuol dire che ben 7 feti su 8 saranno sottoposti ad una terapia senza realmente averne bisogno.
Pertanto, per evitare la somministrazione di desametazone nei maschi o nelle femmine non affette è necessario effettuare una precoce ed accurata diagnosi genetica.
L’efficacia della terapia prenatale sul feto è ancora incerta ma sembra certo, oramai, che la terapia non causi alcuna malformazione congenita e che il numero di feti morti durante la gravidanza non sia superiore a quello della popolazione generale, anche se alcuni effetti collaterali dei glucocorticoidi potrebbero passare inosservati durante le prime fasi della vita fetale.
Alcune complicazioni, invece, potrebbero riguardare la madre; infatti lo sviluppo della sindrome di Cushing, l’incremento ponderale e lo sviluppo di ipertensione sono stati riscontrati nell’1% delle donne trattate in gravidanza.
Sempre per ovviare agli effetti collaterali del farmaco si è pensato di ridurre il dosaggio del desametazone durante la fase finale della gravidanza anche se non sono stati ancora effettuati studi che confermino l’utilità di questo schema a scalare.
In ogni modo è importante informare la madre sugli effetti collaterali del farmaco, sui potenziali rischi che essa stessa corre e sulla possibilità che la terapia possa anche non essere completamente efficace sulle figlie affette.
Comunque si consiglia un adeguato trattamento solo sotto lo stretto controllo da parte di centri specializzati e di personale esperto.
{kind=link}
NUOVE PROSPETTIVE TERAPEUTICHE
Bloccare la sintesi o l’azione degli ormoni sessuali surrenalici permetterebbe di ridurre il dosaggio dei glucocorticoidi.
I risultati preliminari di uno studio in corso evidenziano che l’utilizzo di uno schema con 4 farmaci (idrocortisone a basso dosaggio, fludrocortisone, testolactone (un inibitore dell’aromatasi che dovrebbe impedire la saldatura delle epifisi indotta dagli estrogeni) e flutamide (un antiandrogeno non steroideo che agisce come antagonista sul recettore androgenico prevenendo la virilizzazione) comporta un minor avanzamento dell’età ossea, un più lento incremento ponderale e una ridotta velocità di crescita, se confrontate con quelle dei soggetti trattati con lo schema classico ad alto dosaggio di idrocortisone e fludrocortisone (28). I dati riscontrati sembrano confortanti in tal senso in quanto, a dispetto dei valori più elevati di 17-idrossi progesterone, androstenedione e testosterone riscontrati nei pazienti trattati con lo schema dei quattro farmaci, si è osservata, comunque, una normale maturazione ossea e una normalizzazione della velocità di crescita (28,37).
Durante i primi due anni di follow-up, inoltre, non sono stati riscontrati particolari effetti collaterali dovuti a questo schema terapeutico ad eccezione di una più elevata incidenza di pubertà precoce centrale.
Una volta completato lo studio in corso, questa modalità terapeutica potrà eventualmente essere presa in considerazione nella terapia del deficit di 21 idrossilasi.
Un’altra terapia sperimentale potrebbe essere l’utilizzo di antagonisti del CRH per ridurre l’iperstimolazione del surrene o del carbenoxolone per incrementare i livelli di biodisponibilità di cortisolo. Gli antagonisti del CRH, in combinazione con i glicocorticoidi e i mineralcorticoidi, potrebbe, infatti, ovviare al trattamento con inibitori dell’aromatasi o alla surrenectomia bilaterale.
L’antalarmin è un prototipo di antagonista del recettore del CRH di tipo I che, bloccando il suddetto recettore, riduce, in acuto ed in cronico, la sintesi di ACTH e di cortisolo, senza provocare un’insufficienza surrenalica (29,30,31).
Inoltre, è stato dimostrato che il sistema simpatico-midollare del surrene, è in grado di stimolare la zona reticolare del surrene attraverso la produzione locale di peptidi simili al CRH. E’ probabile, quindi, che l’antalarmin, agendo anche a livello della zona reticolare, possa bloccare la sintesi degli androgeni agendo anche in questa regione e portando, quindi, un ulteriore beneficio terapeutico (32).
In ogni modo, lo studio preclinico in corso con antagonisti del CRH sembra dare risultati promettenti (29,30,31).
Altri possibili interventi terapeutici sono l’utilizzo dell’ormone della crescita per promuovere l’accrescimento e degli analoghi del GnRH per ritardare la comparsa della pubertà e quindi per consentire il raggiungimento di una discreta altezza definitiva.
Nessuna di queste terapie, però, può essere raccomandata, allo stato attuale, come terapia di base. In alternativa alla terapia farmacologia è stata proposta la surrenectomia bilaterale, per via laparoscopica, con terapia steroidea sostitutiva; i fautori di questa modalità terapeutica, infatti, sostengono che sia più difficile trattare l’iperplasia surrenalica congenita che l’ipocorticosurrenalismo (che fra l’altro, se ben sostituito, non interferisce nemmeno con la crescita e con lo sviluppo puberale) (33,34,35); i contrari a questo approccio, invece, sostengono che si tratti di un trattamento troppo aggressivo per una patologia che può essere tranquillamente controllata farmacologicamente e che comporta una serie di rischi quali la chirurgia in sé, l’anestesia e l’irreversibilità. Inoltre la surrenectomia bilaterale stimola lo sviluppo del tessuto surrenalico residuo intratesticolare che è causato dagli elevati livelli di ACTH.
E’ stato riportato, inoltre, che la surrenectomia bilaterale determina una ridotta produzione di DHEA che può avere degli effetti negativi sul tono dell’umore ed incrementare l’astenia.
Va ricordato, però, che la surrenectomia bilaterale è stata proposta come opzione terapeutica solamente nelle forme particolarmente severe di iperplasia surrenalica congenita. Attualmente, infatti, in letteratura, sono presenti solo 5 casi trattati chirurgicamente (di cui uno per via laparoscopica) e sono tutti casi in cui non vi è stato un adeguato controllo con la terapia farmacologia (33,34).
L’indicazione all’intervento di surrenectomia bilaterale è solo per le iperplasie surrenaliche congenite in cui la terapia farmacologia ha fallito, e si effettua, solitamente, in età avanzata; non è indicata una surrenectomia bilaterale precoce entro il primo anno di vita (magari in concomitanza della correzione chirurgica delle anomalie genitali) perché nel primo anno di vita non è semplice effettuare una previsione sulla severità della patologia neanche basandosi sul genotipo, vista la non perfetta corrispondenza fra genotipo e fenotipo (11).
Infine un’altra possibilità terapeutica potrebbe esser data dalla terapia genica.
Quest’ultima consiste nell’introdurre, attraverso un vettore, il gene codificante per l’enzima 21-idrossilasi all’interno del genoma del topo affetto. Il vettore utilizzato fin ora è solitamente un adenovirus capace di codificare la sequenza del gene umano CYP21 (AdCYP21) (6).
L’introduzione intra-surrenalica dell’AdCYP21 nei topi è seguita da una aumentata espressione di CYP21-mRNA, dalla ricomparsa dell’ attività dell’enzima 21-idrossilasi, dalla normalizzazione della morfologia surrenalica e dei livelli di corticosterone (6). Successivamente si osserva anche una normalizzazione delle anomalie della midollare del surrene. Pertanto a differenza della terapia farmacologia, la terapia genica è in grado di correggere sia la funzionalità della corticale che della midollare del surrene (6,36).
L’utilizzo di un adenovirus come vettore non induce nessuna risposta infiammatoria nella ghiandola surrenalica, probabilmente perché l’elevata concentrazione di glucocorticoidi all’interno del surrene protegge dal processo infiammatorio che questi vettori causano, di norma, negli altri tessuti. Pertanto, questi dati, insieme alla positività dei risultati preliminari, rende la ghiandola surrenalica particolarmente adatta alla terapia genica.
In futuro, l’utilizzo di nuovi vettori (retrovirus che sono in grado di integrarsi stabilmente per lungo tempo nel DNA) con promotori specifici per il surrene, dovrebbe aumentare ulteriormente l’efficacia e la durata della terapia genica.
In questo modo la terapia genica potrà essere la definitiva soluzione dell’iperplasia surrenalica congenita, ma per il momento non è ancora clinicamente applicabile.
Prenota una visita specialistica endocrinologica in merito a questo argomento.
Dott. Massimiliano Andrioli
Specialista in Endocrinologia e Malattie del Ricambio
Centro EndocrinologiaOggi, Roma
viale Somalia 33A, Roma
tel/fax 0686391386
cell 3337831426
Studio EndocrinologiaOggi, Lecce
via Ruffano 4, Casarano (Lecce)
tel/fax 0686391386
cell 3337831426
Share
GIU
2011