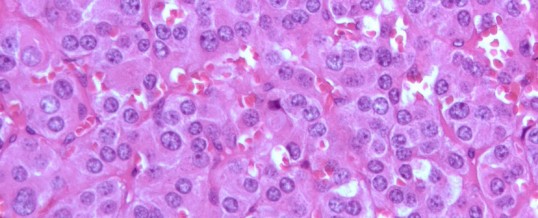
FEOCROMOCITOMA
Il feocromocitoma è un tumore del tessuto cromaffine. Questo tessuto origina dalla cresta neurale e migra localizzandosi nella midollare surrenale e nei gangli delle catene simpatiche nel torace e nell’addome.
In tali sedi può dare origine a tumori che, nella surrene prendono il nome di feocromocitomi ed in sede extrasurrenalica vengono chiamati paragangliomi. Questi tumori sono di origine simpatica ed hanno come caratteristica principale quella di sintetizzare e secernere catecolamine, loro metaboliti e numerose altre sostanze peptidiche e non (1), molte delle quali possiedono proprietà vasoattive (tab. 1).
Con il termine di paraganglioma si indicano tuttavia altri tumori di natura parasimpatica, le cui cellule derivano anch’esse dalla cresta neurale, che sono localizzati nelle regioni della testa, del collo e del torace anteriore. Questi paragangliomi parasimpatici non secernono generalmente catecolamine e si evidenziano solo per un effetto di compressione sulle strutture circostanti.
{kind=link}
EPIDEMIOLOGIA
La prevalenza del feocromocitoma è stimata intorno a 2 casi per milione.
Il tumore costituisce causa di ipertensione arteriosa in circa un paziente su mille.
In accordo con i dati della letteratura (2-4), in uno studio retrospettivo condotto per conto della Società Italiana di Endocrinologia (SIE) su 284 pazienti italiani affetti da feocromocitoma (5), la localizzazione del tumore è risultata per l’89.4% a livello surrenalico, per l’8,5% a livello extrasurrenalico e per il 2,1% contemporaneamente intra ed extrasurrenalico. Quando intrasurrenalico il tumore è risultato bilaterale nell’11% dei casi e monolaterale nell’89%. Fra i monolaterali, la localizzazione nella surrene destra è risultata più frequente che nella sinistra (57 contro 32%, p<0.001).
La localizzazione extrasurrenalica è risultata più frequente in pazienti con età inferiore a 20 anni (17.6%) rispetto a pazienti fra 20 e 60 anni (11.8%) e a pazienti con età superiore ai 60 anni (6.1%).
Il tumore è risultato maligno nel 9.9% dei casi e la malignità è risultata più frequente nei tumori extrasurrenalici (32.1%) rispetto a quelli surrenalici (7.2%).
L’età di insorgenza è la più variabile e non viene riportata prevalenza di sesso. Nello studio SIE l’età variava dagli 8 agli 84 anni e la presenza nel sesso femminile (53.6%) non era dissimile da quella negli uomini (46.4%).
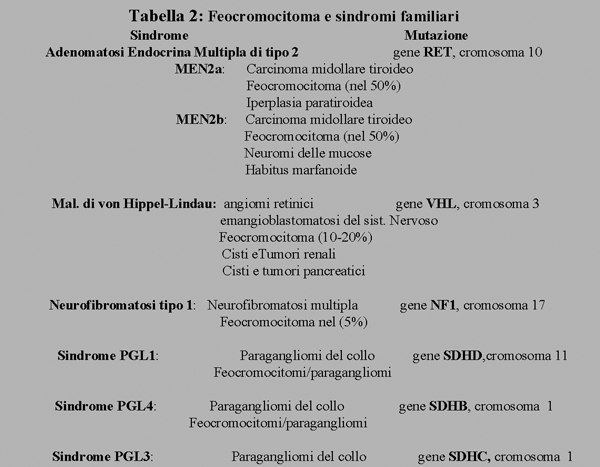
Il tumore si può presentare come sporadico o come componente di sindrome geneticamente trasmessa. Fino a poco tempo fa la familiarità era riportata nel 10% dei casi. Studi recenti hanno invece dimostrato che l’insorgenza del feocromocitoma è legata a mutazioni germinali di vario tipo (tab. 2) nel 25% circa dei casi (6).
{kind=link}
GENETICA
Dati recenti indicano che le forme familiari sono più frequenti di quanto ritenuto finora e che si attestino intorno al 25% delle forme apparentemente sporadiche (6). I geni di suscettibilità per l’insorgenza di feocromocitoma/paraganglioma sono (7): il gene VHL, responsabile della sindrome di von Hippel-Lindau, il gene RET, associato alla Neoplasia Endocrina Multipls di tipo 2 (MEN2), il gene NF-1, associato alla neurofibromatosi di tipo 1 ed i geni che codificano le subunità B, C e D della succinodeidrogenasi (SDH) mitocondriale, associati a sindromi feocromocitoma/paraganglioma denominate PGL4, PGL3 e PGL1 rispettivamente.
In tutte le sindromi suddette, i tumori cromaffini non sono sempre presenti e raramente costituiscono la prima manifestazione clinica. Infatti le forme di feocromocitoma sindromico sono associate con altre neoplasie, sia benigne che maligne.
Nella sindrome VHL (8) il feocromocitoma può essere assente (forma tipo 1), associato ad altre neoplasie (forme tipo 2A e 2B) o costituire la sola manifestazione della sindrome (forma 2C). In media il feocromocitoma è presente nel 10-20% deo pazienti e la età media di insorgenza è intorno ai 30 anni. Nel VHL il feocromocitoma è noradrenalina-secernente (9), è spesso bilaterale, mentre le localizzazioni extrasurrenaliche sono rare e rappresentate da paragangliomi addominali o toracici e molto raramente del collo. Le forme maligne sono rare e rappresentano circa il 5%.
Nella MEN2 il feocromocitoma rappresenta la prima manifestazione clinica nel 25% circa dei casi (10). Si presenta nel 50% dei pazienti, è quasi sempre localizzato nelle surreni, generalmente secerne adrenalina. (e noradrenalina) (9), è molto spesso bilaterale, sebbene le due surreni possano essere interessate in tempi diversi, ed è molto raramente maligno (5%).
Nella NF-1 il feocromocitoma è raro (5%) ed è noradrenalina-secernente.
Nelle sindromi PGL (11) le mutazione dell’SDHC (PGL3) sono molto rare mentre quelle dell’SDHD (PGL1) e dell’SDHB (PGL4) sono state riscontrate in circa il 5% dei pazienti con feocromocitoma apparentemente sporadico (12). Le sindromi PGL sono caratterizzate dalla presenza di paragangliomi del collo e della testa. Questi tumori sono di origine parasimpatica, sono spesso bilaterali o multipli e generalmente non secernenti. Possono associarsi con tumori cromaffini secernenti surrenalici o extrasurrenalici, localizzati nel torace o nell’addome.
La PGL1 e la PGL4 differiscono per diversi aspetti: i paragangliomi della testa e del collo sono più frequenti nella PGL1; la penetranza sembra maggiore nella PGL1 rispetto alla PGL4; la molteplicità è più frequente nella PGL1 che nella PGL4; nella PGL4 i tumori sono maligni con alta frequenza (25-30%) mentre nella PGL1 la malignità è praticamente assente; nella PGL4 sono state segnalati altri tumori maligni come il carcinoma papillifero della tiroide ed il carcinoma renale; infine, il gene SDHD, ma non il gene SDHB, presenta un imprinting materno per cui nella PGL1 solo i pazienti che hanno ereditato il gene mutato dal padre sviluppano la malattia mentre coloro che hanno ereditato la mutazione dalla madre non sono a rischio.
I feocromocitomi/paragangliomi familiari mostrano una età di insorgenza più precoce rispetto agli sporadici e nelle prime due decadi di vita possono costituire il 50-70% dei casi (6). Pertanto, tanto più giovane è il paziente, tanto più è raccomandato il test genetico. Ulteriori indicazioni per il test genetico sono, in aggiunta ad una storia familiare positiva, la presenza di feocromocitomi bilaterali o ricorrenti, la presenza di paragangliomi testa/collo bilaterali o multipli o l’associazione fra feocromocitomi/paragangliomi ed altri tumori maligni.
Il quadro clinico può dare indicazioni su quali test genetici eseguire per primi: La presenza di feocromocitomi bilaterali secernenti adrenalina o l’associazione con carcinoma midollare della tiroide suggerisce l’analisi di RET. Feocromocitomi bilaterali secernenti noradrenalina o la presenza di tumori cromaffini surrenalici ed extrasurrenalici o l’associazione con tumori o cisti renali o con emangioblastomi del SNC, indirizza verso un analisi del VHL. La associazione fra tumori cromaffini e paragangliomi testa/collo suggerisce l’analisi del gene SDHD o SDHB se il feocromocitoma è maligno.
Considerata l’alta frequenza delle forme familiari, diversi Autori suggeriscono di eseguire l’analisi genetica in tutti i casi di feocromocitoma/paraganglioma.
La diagnosi di forma familiare si basa sulla dimostrazione di una mutazione germinale sul DNA dei leucociti circolanti ed ha conseguenze cliniche importanti sia per il paziente che per i suoi familiari. Infatti, poiché le forme sindromiche si associano ad altre neoplasie, sia benigne che maligne, il test genetico può permettere una diagnosi precoce di questi tumori migliorandone la prognosi.
Inoltre, nel paziente con una mutazione germinale, la probabilità di forme multiple o ricorrenti impone un follow-up clinico per tutta la loro vita.
{kind=link}
CLINICA
E’ caratterizzato da una estrema variabilità. La ragione di tale variabilità va ricercata nella diversità quantitativa e qualitativa della secrezione da parte del tumore e nel diverso grado di sensibilità dei tessuti periferici alla azione delle catecolamine causato dal possibile instaurarsi del fenomeno della “desensitizzazione recettoriale”.
In accordo con i dati della letteratura (2-4), anche nella casistica dello studio SIE i sintomi riscontrati con maggior frequenza (tab. 3) erano le palpitazioni (58.1%), la cefalea (51.9%), l’iperidrosi (48.8%) e l’ansia (35.3%). L’associazione di tre di questi sintomi era presente nel 36.5% e la loro contemporanea presenza è stata riscontrata solo nel 15.5% dei casi a conferma della scarsa specificità del quadro sintomatologico e pertanto della grande difficoltà del sospetto clinico. (Prenota una visita endocrinologica).
Anche la valutazione dei parametri cardiovascolari, in ragione della sua variabilità, non offre elementi sicuri per porre diagnosi ma offre solo, e solo un alcuni pazienti, elementi di sospetto. Infatti solo il 59.7% dei pazienti riferiva la presenza della tipica crisi ipertensiva mentre ben il 21.1% dei pazienti presentava una condizione di normotensione sia in clino che in ortostatismo. (Prenota un holter pressorio).
La frequenza di normotensione era poi ancora più alta nei pazienti con feocromocitoma familiare (46%).
Queste caratteristiche spiegano perché nello studio SIE ben l’11.2 % dei feocromocitomi si è presentato come una massa surrenalica incidentale e perché in letteratura sia riportato come molti feocromocitomi siano un reperto autoptico inaspettato (13).
In conclusione, nei pazienti con feocromocitoma il quadro clinico può al massimo presentare elementi di sospetto ma non offre mai certezze diagnostiche che devono essere ricercate solo nei dati di laboratorio. Gli elementi di sospetto derivano dalla presenza di ipertensione, specie se con episodi di parossismo o resistente alla comune terapia antiipertensiva, o dal riscontro di una massa surrenalica.
{kind=link}
DIAGNOSI
Si basa esclusivamente sul riscontro laboratoristico di una eccessiva o alterata secrezione di catecolamine e/o di loro metaboliti. La letteratura è molto ricca di lavori finalizzati a stabilire quale sia il modo migliore per arrivare alla diagnosi (14-26). Le misure possono essere effettuate sia nel sangue che nelle urine e i composti più comunemente misurati sono le catecolamine stesse (noradrenalina e adrenalina), le metanefrine (metanefrina e normetanefrina), l’acido vanilmandelico.
Lavori recenti indicano nella misura delle metanefrine frazionate plasmatiche o urinarie il mezzo più sensibile per la diagnosi di feocromocitoma (22, 23, 26). La sensibilità della misura è da ritenere il dato più importante in questo processo diagnostico dove, per la pericolosità del tumore, ciò che va evitato è il risultato falso-negativo. La misura delle metanefrine comporta un certo numero di falsi positivi ma, a parità di specificità con gli altri metaboliti, resta comunque la misura più sensibile (26). (Prenota una visita endocrinologica).
Pertanto, potendo scegliere fra i vari dosaggi, quello da raccomandare nel sospetto di feocromocitoma è la misura delle metanefrine. Questa misura non è diffusa nei laboratori, specie italiani, dove il metodo più comunemente impiegato è quello dell’acido vanilmandelico (VMA) nelle urine. Questa misura offre un’alta specificità, specie se la metodica impiegata è quella con cromatografia liquida ad alta pressione (HPLC), ma è anche quella che comporta il maggior numero di falsi negativi (bassa sensibilità).
La misura delle catecolamine sia nel plasma che nelle urine, offre caratteristiche di sensibilità e specificità intermedie rispetto alle metanefrine ed al VMA. La misura acquista maggior valore diagnostico quando il risultato del dosaggio viene interpretato alla luce della condizione clinica del paziente al momento del prelievo o della raccolta urinaria. Infatti spesso il feocromocitoma secerne in modo discontinuo e pertanto il dato di laboratorio può risultare diverso a seconda del momento secretorio. In questo senso è fondamentale valutare se nel periodo della raccolta urinaria o al momento del prelievo il paziente presenta normo- o ipertensione, è sintomatico o meno. Ad esempio, il riscontro di normali valori di catecolamine plasmatiche in un campione prelevato in corso di crisi ipertensiva esclude virtualmente la presenza di feocromocitoma.
Il vantaggio della misura delle metanefrine risiede nel fatto che queste vengono sempre secrete dal tumore in modo continuo con meccanismo diverso e autonomo rispetto a quello delle catecolamine (27) che invece è spesso di tipo intermittente.
Uno dei vantaggi offerti dal dosaggio delle metanefrine è che un riscontro di normali valori di metanefrina e normetanefrina nel plasma o nelle urine permette in pratica di escludere la presenza del feocromocitoma. Il riscontro di valori elevati assicura della presenza del tumore solo se l’aumento è superiore ad un certo livello che, nella nostra esperienza, per la normetanefrina plasmatica è di 2.19 nmol/L e per la metanefrina plasmatica è di 0.73 nmol/L (26).
Aumenti delle concentrazioni al di sotto di questi limiti sono da considerare dubbi e pertanto necessitano di ulteriori indagini di approfondimento che vanno dalla ripetizione delle misure basali, all’uso di test dinamici . Fra questi il test di stimolo con glucagone (28) va considerato obsoleto per la sua scarsa sensibilità e specificità e soprattutto per la sua pericolosità. Al contrario il test di soppressione con clonidina (29) è un test non pericoloso, che può risultare utile per meglio definire la diagnosi nei pazienti con aumenti borderline delle catecolamine o delle metanefrine. (Prenota una visita endocrinologica).
Il test si basa sul principio che una attivazione cronica del sistema simpatico, che comporta un aumento del rilascio di catecolamine e un aumento delle metanefrine per loro metabolizzazione periferica, risente, a differenza della secrezione tumorale, dell’azione di inibizione esercitata dalla clonidina attraverso lo stimolo dei recettori alfa-2 presinaptici. Ne deriva che aumentati livelli di catecolamine causati da attivazione simpato-adrenergica vengono riportati a valori normali dalla clonidina che invece non esercita alcun effetto su livelli aumentati per secrezione tumorale.
Il test prevede la misura delle catecolamine o delle metanefrine plasmatiche, è facilmente praticabile ed ha poche controindicazioni. Fra queste la sua esecuzione in pazienti in trattamento con beta-bloccanti nei quali la clonidina potrebbe indurre eccessiva bradicardia o brachiaritmie.
Inoltre occorre ricordare che il test non può essere eseguito in pazienti in trattamento con antidepressivi triciclici, nei quali, fra l’altro, i valori delle catecolamine e delle metanefrine risultano spesso superiori alla norma. Infatti gli antidepressivi triciclici, attraverso l’inibizione del reuptake neuronale, aumentano i livelli intersinaptici di noradrenalina e causano una down regulation dei recettori alfa-2 presinaptici riducendo pertanto l’azione della clonidina. In tali pazienti il test offre risposte falso-positive. Il periodo di sospensione degli antidepressivi necessario per ristabilire la sensibilità alla clonidina è piuttosto lungo e si aggira intorno alle tre, quattro settimane.
In conclusione, per la diagnosi di feocromocitoma, le misure da previlegiare sono quelle delle metanefrine frazionate plasmatiche o urinarie. Le catecolamine plasmatiche offrono buone indicazioni specie se i risultati vengono interpretati alla luce delle condizioni cliniche del paziente al momento del prelievo o della raccolta urinaria. I test dinamici sono da riservare a casi molto selezionati e vanno limitati a quello di inibizione con clonidina.
LOCALIZZAZIONE
Il feocromocitoma si localizza nel 90% dei casi nella surrene. Se il laboratorio mostra una alterazione della adrenalina o della metanefrina, la patologia è certamente a carico della midollare surrenale.
L’indagine di prima scelta è pertanto la TC che permette una ottima definizione dei caratteri anatomici dei surreni (30). Anche la RMN fornisce accurate informazioni circa eventuali lesioni espansive a carico dei surreni. Essa inoltre può dare suggerimenti circa la natura cromaffine dell’eventuale lesione surrenalica che appare iperintensa nelle scansioni T2 pesate (31).
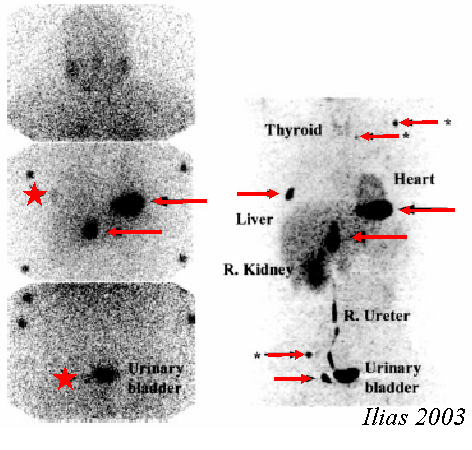
In ogni caso informazioni certe circa la natura cromaffine della neoplasia vengono fornite dalla scintigrafia con MIBG marcata con iodio radioattivo (32). Il tracciante si concentra nelle cellule cromaffini con un meccanismo simile a quello dell’uptake-1 neuronale ed è capace di evidenziare feocromocitomi intra ed extrasurrenalici ed anche lesioni metastatiche.
Per tali ragioni gli esami radiologici (TC/RMN) e la scintigrafia sono da considerarsi complementari (33) e vanno eseguiti entrambi in fase prechirurgica.
La scintigrafia con MIBG, che ha rappresentato un avanzamento fondamentale nella diagnostica di localizzazione del feocromocitoma, presenta una sensibilità intorno al 90% (10% di falsi negativi).
L’uso alternativo di altri traccianti, come la octreotide (34) non si è dimostrato superiore ed anzi causa un numero maggior di falsi negativi (35).
In anni recenti si è cominciato ad utilizzare anche la PET (36), facendo ricorso all’utilizzo di vari traccianti fra cui il 2-desossiglucosio (37), l’idrossiefedrina (38), l’epinefrina (39), la fluorodopamina (40). Questa ultima si è dimostrata capace di evidenziare tumori che erano risultati negativi alla scintigrafia con MIBG (41).
In quei rari casi di feocromocitoma, quasi sempre a sede extrasurrenale, in cui sia le immagini radiologiche che la scintigrafia hanno dato esito negativo, la localizzazione del tumore può essere effettuata attraverso un cateterismo dell’albero venoso con prelievi di plasma a vari livelli per la ricerca del gradiente secretorio evidenziabile dalla misura delle catecolamine plasmatiche.
Nella nostra esperienza tale metodica si è rivelata quasi sempre utile e decisiva per localizzare il feocromocitoma. I risultati migliori si ottengono nei pazienti con feocromocitomi che secernono in modo costante e non “a crisi”. In ogni caso durante il cateterismo occorre monitorare le condizioni cardiocircolatorie del paziente perché il sopravvenire di una crisi ipertensiva (e secretoria) può, se non accuratamente registrata, simulare un gradiente che è falso.
Eventuali dosaggi su prelievi effettuati in modo selettivo nelle vene surrenaliche vanno interpretati con molta cautela per quanto riguarda le concentrazioni assolute di catecolamine che possono essere estremamente variabili dipendendo dalla fase secretiva delle surreni e dal flusso ematico della vena. L’unico dato clinicamente rilevante nei prelievi selettivi dalle vene surrenaliche è il rapporto di concentrazione fra adrenalina e noradrenalina. Tale rapporto nelle surreni normali è pressoché costante intorno a 4, indipendentemente dalla fase secretiva (42). Un rapporto invertito con valori di noradrenalina superiori a quelli di adrenalina indica una condizione di patologia.
{kind=link}
TERAPIA
La terapia del feocromocitoma è chirurgica ed è risolutiva nei casi benigni. (Prenota una visita chirurgica).
Per i feocromocitomi a localizzazione surrenalica la via chirurgica elettiva di approccio è quella laparoscopica (43).
La terapia medica è di importanza capitale per la preparazione del paziente all’intervento chirurgico, per il trattamento delle complicanze operatorie e per la terapia delle forme maligne o comunque non operabili (44).
Il cardine della terapia medica è l’uso di farmaci ad azione alfa-bloccante (45).
Essi non interferiscono sul rilascio delle catecolamine da parte del tumore ma ne limitano gli effetti periferici bloccando l’azione delle catecolamine sui recettori vascolari alfa vasocostrittori. Inducono pertanto una vasodilatazione ed una conseguente normalizzazione della pressione arteriosa. Essi inoltre permettono una riespansione del volume circolante, che è ridotto nei pazienti con feocromocitoma e annullano la down regulation dei recettori alfa vascolari determinata dalla loro eccessiva esposizione agli alti livelli di catecolamine. Questi effetti sono fondamentali per impedire la temibile e talora irreversibile crisi ipotensiva che può seguire all’asportazione del tumore.
Fra gli alfa antagonisti si distinguono quelli non selettivi (alfa-1 e alfa-2 antagonisti) come la fenossibenzamina e quelli selettivi (alfa-1 antagonisti) come la doxazosina.
La fenossibenzamina è un inibitore non competitivo, ha una durata d’azione di circa 24 ore, è somministrabile per os e comporta un blocco, oltre che degli alfa recettori postsinaptici vascolari (alfa-1) anche di quelli pre-sinaptici (alfa-2). Il blocco di questi ultimi comporta un aumentato rilascio di noradrenalina dai terminali nervosi simpatici con conseguente tachicardia che quasi sempre causa la necessità dell’aggiunta in terapia di un beta-bloccante. Va ricordato che per nessuna ragione nel paziente con feocromocitoma vanno usati i betabloccanti per ridurre la tachicardia indotta dalle catecolamine tumorali prima di un adeguato alfa-blocco. Infatti un blocco dei recettori beta vasodilatanti, non controbilanciato dal blocco degli alfa, vasocostrittori, può comportare l’aggravamento di una crisi ipertensiva indotta da un improvviso rilascio di catecolamine tumorali.
In corso di improvvisa crisi ipertensiva, se disponibile, il farmaco di scelta è la fentolamina che va somministrata per via endovenosa, diluita 1:10, lentamente, monitorando la pressione arteriosa. Una somministrazione rapida in bolo può infatti determinare una crisi ipotensiva grave. In assenza della fentolamina possono essere utilizzati altri farmaci ipotensivanti come l’urapidil, somministrabile per via endovenosa, o la nifedipina, somministrabile per os.
La doxazosina è un inibitore competitivi e pertanto, pur avendo una emivita simile a quella della fenossibenzamina (circa 20 ore) può essere spiazzata dai recettori da alti livelli circolanti di catecolamine tumorali. Ne consegue che è raccomandabile somministrare il farmaco in più dosi giornaliere. Le dosi di alfa bloccanti vanno progressivamente aumentate fino al raggiungimento, prima della chirurgia, di una condizione pressoché continua di normotensione.
Nei casi di pazienti con feocromocitoma maligno e pertanto non eradicabile chirurgicamente, come in quelli con feocromocitoma benigno ma non operabile per gravi patologie concomitanti, la terapia medica svolge un ruolo fondamentale nel limitare gli effetti dannosi delle catecolamine sul sistema cardiovascolare. La sopravvivenza di questi pazienti è spesso lunga anche di molti anni perché generalmente la progressione delle lesioni è lenta e la prognosi è pertanto fortemente condizionata dagli effetti dannosi delle catecolamine. Inoltre il tumore è poco sensibile sia agli agenti chemioterapici sia alla terapia radiometabolica e pertanto la terapia anticatecolaminergica riveste una importanza primaria nella gestione del paziente con feocromocitoma maligno.
Oltre all’uso degli alfa-bloccanti, in questi pazienti è stata impiegata l’alfa-metil-paratirosina (46). Questa sostanza limita la sintesi delle catecolamine attraverso una inibizione competitiva con la tirosinoidrossilasi che è l’enzima limitante la sintesi delle catecolamine. I risultati clinici sono evidenti ma a spese di effetti collaterali importanti, come l’ipotensione ortostatica, poiché l’inibizione della sintesi non avviene solo a livello del tessuto cromaffine neoplastico, ma anche a livello dei neuroni simpatici.
FOLLOW-UP
In fase postoperatoria è necessario valutare l’andamento della pressione arteriosa e della glicemia e, a quattro settimane dall’intervento, i valori di catecolamine o dei loro metaboliti.
I pazienti con feocromocitoma sporadico che mostrano una normalizzazione dei parametri di laboratorio possono essere considerati guariti ma vanno informati che esiste la possibilità che il tumore possa recidivare o presentare diffusione metastatica anche dopo molti anni dall’intervento. Va pertanto consigliato di effettuare periodici controlli delle metanefrine urinarie con cadenza annuale o all’insorgere di una sintomatologia sospetta.
Nello studio SIE è stato considerato l’effetto dell’intervento chirurgico sulla pressione arteriosa. Su 108 pazienti affetti da feocromocitoma sporadico è stato visto che nel:
– 59.3% (64 pz) si ha normalizzazione della pressione arteriosa;
– 26.8% (29 pz) il paziente resta iperteso ma con riduzione della pressione arteriosa;
– 13.9% (15 pz) il paziente resta iperteso senza riduzione della pressione arteriosa.
Negli ultimi due gruppi la persistenza di valori elevati di pressione arteriosa è da ascrivere o a un’ipertensione essenziale preesistente o a un danno vascolare irreversibile causato dalle catecolamine (ipertrofia della muscolare dei vasi).
Prof. Massimo Mannelli
Bibliografia
1) Manger WM, Gifford RWJ: Phaeochromocytoma. New York Springer-Verlag, 1977.
2) Sever PS, Roberts JC, Snell ME: Phaeochromocytoma. Clin Endocrinol Metab 1980; 9:543-568.
3) Bravo EL, Gifford RW Jr.: Pheochromocytoma: diagnosis, localization and management. N Engl J Med 1984; 311: 1298-1303
4) Cryer PE: Phaeochromocytoma. Clin Endocrinol Metab 1985; 14: 203-220.
5) Mannelli M, Ianni L, Cilotti A, Conti A: Pheochromocytoma in Italy: a multicentric retrospective study. Eur J Endocrinol 1999; 141: 619-24.
6) Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, et al.: Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346: 1486-8.
7) Maher ER, Eng C. The pressure rises: update on the genetics of phaeochromocytoma. Hum Mol Genet. 2002, 11: 2347-54.
8)Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, Oldfield EH. von Hippel-Lindau disease. Lancet 2003, 361:2059-67.
9) G. Eisenhofer, MM Walther, TT Huynh, S Li, SR Bornstein, M. Mannelli, DS Goldstein, A Vortmeyer, JWM Lenders, WMLinehan, K Pacak. Pheochromocytomas in von-Hippel Lindau disease and Multiple Endocrine Neoplasia Type 2 display distinct noradrenergic and adrenergic phenotypes. J Clin Endocrinol Metab 2001, 86: 1999-2008.
10) Modigliani E, Vasen HM, Raue K, Dralle H, Frilling A, Gheri RG, Brandi ML, Limbert E, Niederle B, Forgas L, et al. Pheochromocytoma in multiple endocrine neoplasia type 2: European study. The Euromen Study Group. J Intern Med 1995, ;238: 363-7.
11) Baysal BE, Willett-Brozick JE, Lawrence EC, Drovdlic CM, Savul SA, McLeod DR, Yee HA, Brackmann DE, Slattery WH 3rd, Myers EN, Ferrell RE, Rubinstein WS. Prevalence of SDHB, SDHC, and SDHD germline mutations in clinic patients with head and neck paragangliomas. J Med Genet 2002, 39: 178-83.
12) Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, Buchta M, Franke G, Klisch J, Bley TA, Hoegerle S, Boedeker CC, Opocher G, Schipper J, Januszewicz A, Eng C; European-American Paraganglioma Study Group. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 25;292: 943-51.
13) Bittar DA: Unsuspected phaeochromocytoma. Can Anaest Soc J 1982; 29:183-184.
14) Bravo EL: Evolving concepts in the pathophysiology, diagnosis, and treatment of pheochromocytoma. Endocr Rev 1994; 15: 356-68.
15) Manger WM, Gifford RWJr.: Pheochromocytoma: current diagnosis and management. Clev Clin J Med 1993; 60: 365-78.
16) Mannelli M: Diagnostic problems in pheochromocytoma. J Endocrinol Invest 1989; 12: 739-57.
17) Bravo EL, Tarazi RC, Gifford RW, Stewart BH: Circulating and urinary catecholamines in pheochromocytoma. Diagnostic and pathophysiologic implications. N Engl J Med 1979; 301: 682-86.
18) Duncan MW, Compton P, Lazarus L, Smythe GA: Measurement of norepinephrine and 3,4-dihydroxyphenylglycol in urine and plasma for the diagnosis of pheochromocytoma. N Engl J Med 1988; 319: 136-42.
19) Young MJ, Dmuchowski C, Wallis JW, Barnas GP, Shapiro B: Biochemical tests for pheochromocytoma: strategies in hypertensive patients. J Gen Intern Med 1989; 4: 273-276.
20) Peaston RT, Lai LC: Biochemical detection of phaechromocytoma: Should we still be measuring urinary HMMA? J Clin Pathol 1993; 46: 734-37.
21) Gerlo EA, Sevens C: Urinary and plasma catecholamines and urinary catecholamine metabolites in pheochromocytoma: diagnostic value in 19 cases. Clin Chem 1994; 40: 250-56.
22) Lenders JW, Keiser HR, Goldstein DS, et al.: Plasma metanephrines in the diagnosis of pheochromocytoma. Ann Intern Med 1995; 123: 101-09.
23) Heron E, Chatellier G, Billaud E, Foos E, Plouin PF: The urinary metanephrine-to-creatinine ratio for the diagnosis of pheochromocytoma. Ann Intern Med 1996; 125: 300-03.
24) Witteles RM, Kaplan EL, Roizen MF: Sensitivity of diagnostic and localization tests for pheochromocytoma in clinical practice. Arch Intern Med 2000; 160: 2521-524.
25) Mannelli M, De Feo ML, Maggi M, Pupilli C, Opocher G, Valenza T, Baldi E, Serio M: Usefulness of basal catecholamine plasma levels and clonidine suppression test in the diagnosis of pheochromocytoma. J Endocrinol Invest 1987; 10:377-382.
26) JW Lenders, K Pacak, MM Walther, WM Linehan, M Mannelli, P Friberg, HR kaiser, DS Goldstein, G Eisenhofer: Biochemical diagnosis of pheochromocytoma: which test is best? JAMA 2002; 287: 1427-1434.
27) Eisenhofer G, Keiser H, Friberg P, et al.: Plasma metanephrines are markers of pheochromocytoma produced by catechol-O-methyltransferase within tumors. J Clin Endocrinol Metab 1998; 83: 2175-2185.
28) Lawrance AM: Glucagon provocative test for pheochromocytoma.. Ann Int Med 1967; 66:1091-1096.
29) Bravo EL, Tarazi RC, Fouad FM, Vidt DG, Gifford RWj: Clonidine suppression test: A useful aid in the diagnosis of pheochromocytoma. N Engl J Med 1981; 305: 623-626.
30) Welch TJ, Sheedy PF, Van Heerden JA, Sheps SG, Hattery RR, Stephens DM.: Pheochromocytoma: value of computed tomography. 1983. Radiology 184:501-503.
31) Francis IR, Glazer GM, Shapiro B, Sisson JC, Gross BH: Complementary roles of CT and I131-MIBG scintigraphy in diagnosing pheochromocytoma. 1983. AJR 141:719-725.
32) Schmedtje JFj, Sax S, Pool JL, Goldfarb RA, Nelson EB: Localization of ectopic phaeochromocytomas by magnetic resonance imaging. 1987. Am J Med 83: 770-772.
33) Sisson JC, Frager MS, Valk TW, Gross MD, Swanson DP, Wieland DM, Tobes MC, Beierwaltes WH, Thompson NW: Scintigraphic localization of phaeochromocytoma. N Engl J Med 1981; 305:12-17.
34) Kwekkeboom DJ, van Hurk H, Pauw BK, et al.: Octreotide scintigraphy for the detection of paragangliomas. J Nucl Med 1993; 34: 873-878.
35) van den Harst, deHerder WW, Bruining HA, et al.: 123-I-Metaiodobezylguanidine and 111-In-Octreotide uptake in benign and malignant pheochromocytomas. J Clin Endocrinol Metab 2001; 86: 685-693.
36) Tewson TJ, Krohn KA: PET radiopharmaceuticals: state-of-the-art and future prospects. Semin Nucl Med 1998; 28:221-234.
37) Shulkin BL, Thompson NW, Shapiro B, Francio IR, Sisson JC: Pheochromocytomas: imaging with 2-deoxy-D-glucose PET. Radiology 1999; 212: 35-41.
38) Shulkin BL, Wieland DM, Schwaiger M, Thompson NW, Francis IR, Haka MS, et al.: PET scanning with hydroxyephedrine: an approach to the localization of pheochromocytoma. J Nucl Med 1992; 33: 1125-1131.
39) Shulkin B: PET epinephrine studies of pheochromocytoma. J Nucl Med 1995; 36: 22P-23P.
40) Hovevey-Sion D, Heisenhofer G, Kopin IJ, Kirk KL, Chang PC, Szemeredi K, et al.: Metabolic fate of injected radiolabelled dopamine and 2-fluorodopamine in rats. Neuropharmacology 1990; 29: 881 887.
41) Pacak K, Linehan WM, Eisenhofer G, Walter MM, Goldstein DS: Recent advances in genetics, diagnosis and treatment of pheochromocytoma. Ann Int Med 2001; 134: 315-329.
42) M. Mannelli, R.G. Gheri, C. Selli, D. Turini, A. Pampanini, G. Giusti, M. Serio: A study of human adrenal secretion. Measurement of epinephrine, norepinephrine, dopamine and cortisol in peripheral and adreanl venous blood under surgical stress. J Endocrinol Invest 1982; 5: 91-95.
43) Gagner M, Lacroix A, Prinz RA, et al. : Early experience with laparoscopic approach for adrenalectomy. Surgery 1993; 114: 1120-1125.
44) Fonseca V, Bouloux PMG: Phaeochromocytoma and paraganglioma.
Baillieres Clin Endocrinol Metab 1993; 7:509-44.
45) Mannelli M. Management and Treatment of Pheochromocytomas and Paragangliomas Proc N Y Acad Sci 2006 (in press)
46) Engelman K, Hoewitz D, Jequier E, et al.: Biochemical and pharmacological effects of alpha-metyltyrosine in man. J Clin Invest 1968; 47: 577-594.
GIU
2011